一種固定三聯吡啶釕及電化學發光檢測亮藍的方法與流程
本發明屬于電化學分析測試技術領域,具體涉及一種以聚磺基水楊酸—三聯吡啶釕修飾玻碳電極為工作電極,定量檢測亮藍的電化學發光方法。
背景技術:
亮藍,又名食用藍色一號,屬人工合成色素,為非偶氮類色素,是由苯甲醛鄰磺酸與N-乙基-N-(3-磺基芐基)-苯胺經縮合、氧化而制得。作為一種現代食品工業常用的人工合成色素,為保證公共健康,我國《食品添加劑使用衛生標準》規定:亮藍在食品中的最大使用限量為0.025g/kg。目前國內外有關亮藍的檢測技術主要有高效液相色譜法、熒光光譜法以及分光光度法等,這些方法檢測精度雖高,但樣品前處理復雜,儀器昂貴,需要專業的操作人員,耗時長。電化學發光法以其靈敏度高、操作簡便和成本低等優點,成為檢測亮藍的新方法。使用合適的化學修飾電極,對于提高亮藍檢測的選擇性、靈敏度和重現性等主要性能,具有重要的現實意義和學術價值。
技術實現要素:
本發明要解決的技術問題是,針對現有食品中亮藍的檢測方法大都需要大型的實驗儀器且實驗操作條件苛刻,需專業人員操作,樣品前處理復雜、成本高等缺點,提供一種簡單、快速、靈敏度高、選擇性好的檢測亮藍的電化學發光法。
本發明解決其技術問題所采用的技術方案是,一種檢測亮藍的電化學發光法,以聚磺基水楊酸—三聯吡啶釕修飾玻碳電極為工作電極,飽和甘汞電極作為參比電極,鉑電極作為輔助電極,組成三電極體系進行檢測。進一步地,上述檢測方法具體步驟如下:
(1)聚磺基水楊酸—三聯吡啶釕修飾玻碳電極的制備:
配制磺基水楊酸、三聯吡啶釕混合電聚合溶液,將玻碳電極置于電聚合溶液中循環伏安掃描10-30圈,掃描范圍:0.1-1.5V;掃速:0.1V/s,電聚合完成后沖洗干燥以作為電化學發光測試的工作電極;
(2)標準曲線的繪制:
將步驟(1)制備的修飾玻碳電極作為工作電極,飽和甘汞電極作為參比電極,鉑電極作為輔助電極,組成三電極體系;將該三電極體系置于含有不同濃度亮藍的緩沖溶液中,在0.1V到1.5V的電位范圍內進行循環伏安法掃描,記錄電位-發光強度(E-ECL)曲線,并讀出發光強度峰值。所得數據經統計后,以亮藍的濃度為橫坐標,空白溶液和亮藍溶液的發光強度差值為縱坐標,繪制標準曲線,進而獲得相應的線性回歸方程。
(3)樣品的檢測:
將樣品用緩沖溶液進行溶解后過濾,按照與步驟(2)相同的電化學發光測試方法對待測樣品溶液進行測試,以獲得發光強度峰值,所得發光強度峰值用上述步驟(2)所得標準曲線所對應的線性回歸方程計算出待檢測樣品中的亮藍濃度。
作為優選,所述電聚合液中含磺基水楊酸濃度為0.01mol/L,三聯吡啶釕濃度為0.1mmol/L。所述循環伏安法掃速為0.1V/s,所述的緩沖溶液為pH=10的0.1mol/L的磷酸緩沖溶液(PBS)。
本發明的有益效果是:本發明采用聚磺基水楊酸來固定三聯吡啶釕,利用磺基水楊酸與三聯吡啶釕之間存在的氫鍵作用,特別是磺基水楊酸聚合成聚磺基水楊酸時與三聯吡啶釕的氫鍵作用會更強,使得磺基水楊酸電聚合膜能夠很好的固定三聯吡啶釕得到修飾電極,具有優異的吸附亮藍性能,通過檢測不同亮藍濃度下修飾電極的電化學發光強度,選擇性檢測亮藍。該電極對亮藍檢測的線性范圍為5.0×10-7~1.0×10-5mol/L,檢測限為1.0×10-7mol/L。與其它檢測方法相比,其簡單快速、易操作,用于實際樣品薄荷糖中亮藍的測定,本測試方法具有較高的靈敏度、較好的選擇性。
附圖說明
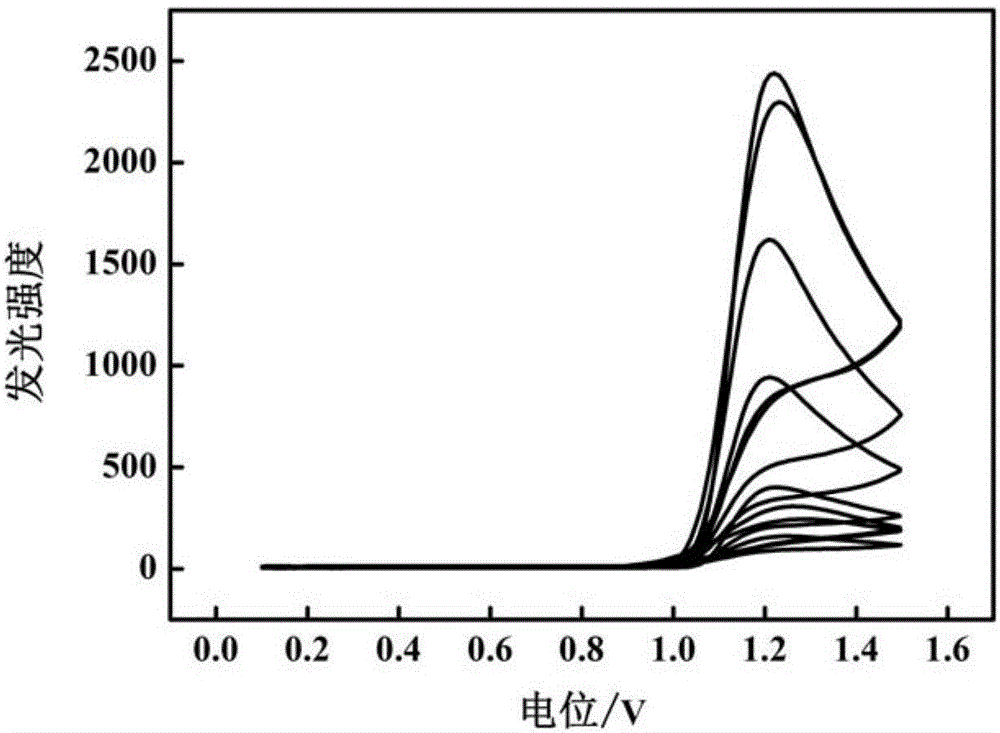
圖1是聚磺基水楊酸—三聯吡啶釕修飾玻碳電極在不同濃度亮藍的PBS緩沖溶液中檢測的E-ECL曲線,其中亮藍的濃度按曲線峰值高低從上到下依次為:5.0×10-7mol/L,1.0×10-6mol/L,3.0×10-6mol/L,5.0×10-6mol/L,7.0×10-6mol/L,8.0×10-6mol/L,9.0×10-6mol/L和1.0×10-5mol/L。
圖2是亮藍溶液對比空白溶液發光強度差值與對應濃度的標準曲線。
具體實施方式
本發明下面結合實施例作進一步詳述:
實施例1:
(1)磺基水楊酸—三聯吡啶釕修飾電極的制備:
將玻碳電極置于含0.01mol/L磺基水楊酸與0.1mmol/L三聯吡啶釕的混合電聚合溶液中,采用循環伏安法進行電聚合,在-1.5~+2.0V的電位范圍內,掃速0.1V/s,掃描30圈,取出后用去離子水沖洗晾干,得到修飾電極;
(2)標準曲線的繪制:
將前述聚磺基水楊酸—三聯吡啶釕修飾玻碳電極作為工作電極,飽和甘汞電極作為參比電極,鉑電極作為輔助電極,組成三電極體系;將該三電極體系置于含有不同濃度亮藍(5.0×10-7mol/L,1.0×10-6mol/L,3.0×10-6mol/L,5.0×10-6mol/L,7.0×10-6mol/L,8.0×10-6mol/L,9.0×10-6mol/L和1.0×10-5mol/L)的0.1mol/L pH=10的PBS緩沖溶液中,在0.1V到1.5V的電位范圍內進行循環伏安法掃描。參數設定為掃速0.1V/s,在此條件下,記錄E-ECL曲線,并讀出發光強度峰值,如圖1所示。以亮藍的濃度為橫坐標,空白溶液與亮藍溶液發光強度差值為縱坐標,得到標準曲線,該標準曲線的線性分為兩段,其線性回歸方程分別為:ΔECL1=82.9C(×10-6mol/L)+2014(5.0×10-7mol/L~7.0×10-6mol/L)(線性相關系數R=0.990);ΔECL2=319.96C(×10-6mol/L)+370.93(7.0×10-6mol/L~1.0×10-5mol/L)(線性相關系數R=0.990),其中C為亮藍的濃度,ΔECL為空白溶液和亮藍溶液發光強度差值(如說明書附圖2),檢測限為1.0×10-7mol/L。并利用標準曲線法對食品中亮藍進行分析檢測。
(3)樣品的檢測
選取市售的薄荷糖試樣兩粒放在燒杯中,加pH=10的0.1mol/L PBS溶解,用0.22μm的濾膜過濾后,倒入50mL容量瓶中定容,取20mL所得溶液用于電化學發光檢測;按照與步驟(2)相同的電化學測試方法對待測樣品溶液進行測試,以獲得ECL發光強度值及其與空白溶液發光強度值差值,所得發光強度值差值用步驟(2)所得標準曲線所對應的線性回歸方程計算出待檢測樣品中亮藍的濃度。
測定結果表明:測得的ECL發光強度值為2380,即所測樣品中含有亮藍7.78×10-7mol/L。對樣品進行加標回收,計算加標回收率,檢測結果如表1所示。
對比實施例1:
(1)三聯吡啶釕修飾玻碳電極的制備
將玻碳電極置于0.1mmol/L三聯吡啶釕的混合電聚合溶液中,采用循環伏安法進行電聚合,在-1.5~+2.0V的電位范圍內,掃速0.1V/s,掃描30圈,取出后用去離子水沖洗晾干,得到修飾電極。
(2)標準曲線的繪制:
將步驟(1)得到的三聯吡啶釕修飾玻碳電極作為工作電極,飽和甘汞電極作為參比電極,鉑電極作為輔助電極,組成三電極體系;將該三電極體系置于含有不同濃度亮藍(5.0×10-7mol/L,1.0×10-6mol/L,3.0×10-6mol/L,5.0×10-6mol/L,7.0×10-6mol/L,8.0×10-6mol/L,9.0×10-6mol/L和1.0×10-5mol/L)的0.1mol/L pH=10的PBS緩沖溶液中在0.1V到1.5V的電位范圍內進行循環伏安法掃描。參數設定為掃速0.1V/s,在此條件下,記錄E-ECL曲線,并讀出發光強度峰值,以亮藍的濃度為橫坐標,空白溶液與亮藍溶液發光強度差值為縱坐標,得到標準曲線,進而獲得相應的線性回歸方程。
(3)樣品的檢測
選取市售的薄荷糖試樣兩粒于燒杯中,加pH=10的0.1mol/L PBS溶解,用0.22μm的濾膜過濾后,倒入50mL容量瓶中定容,取20mL所得溶液用于電化學發光檢測;按照與步驟(2)相同的電化學測試方法對待測樣品溶液進行測試,以獲得ECL發光強度值及其與空白溶液發光強度值差值,所得發光強度值差值用步驟(2)所得標準曲線所對應的線性回歸方程計算出待檢測樣品中亮藍的濃度。
表1樣品加標回收測定結果
加標回收率在101.4%~103.7%,說明該方法具有很好的準確性。
基于上述結果,本發明的電化學發光法檢測亮藍能同樣達到較低的檢測限和較高的靈敏度,能夠對樣品中亮藍進行準確的定量分析。
上述較佳實施例僅用于說明本發明的內容,但這并非是對本發明的限制,本領域的相關技術人員,在不脫離本發明的范圍的情況下,還可以做出相應的調整和變型,因此所有等同替換或等效變型的方式形成的技術方案均屬于本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!