通過配體交換的納米晶體的能級修飾的制作方法
本申請要求于2014年5月9日提交的美國臨時申請No.61/990,789的優先權,其通過引用以其整體并入。
技術領域
本發明涉及半導體納米晶體(semiconductor nanocrystal)。
背景技術:
其半徑小于整體(主體,bulk)激發玻爾半徑的半導體納米晶體(量子點)構成一類介于分子和大塊形式的物質之間的材料。電子和空穴兩者在整個三維中的量子約束導致該材料的有效帶隙隨著晶粒尺寸減小而增大。半導體納米晶體已經成為受到極大關注的、有前途的廣泛應用的主題,該應用包括顯示器件、信息存儲、生物標記材料、光伏器件、傳感器和催化劑。
技術實現要素:
一方面,改善光伏器件的性能的方法可包括通過配體交換修飾(modify,修正,更改)所述器件的半導體納米晶體的表面能級。在某些實施方式中,所述配體可包括硫醇。在某些實施方式中,所述配體可包括胺。
在某些實施方式中,所述配體可包括鹵化物。
在某些實施方式中,所述半導體納米晶體可包括硫化鉛。
在某些實施方式中,所述配體可包括苯硫酚(BT)、1,2-苯二硫酚、1,3-苯二硫酚、1,4-苯二硫酚、1,2-乙烷二硫醇或3-巰基丙酸。在某些實施方式中,所述配體可包括1,2-乙二胺或硫氰酸銨。在某些實施方式中,所述配體可包括碘化四丁基銨、溴化四丁基銨、氯化四丁基銨或氟化四丁基銨。
另一方面,具有改善的性能的光伏器件可包括:第一電極;與第一電極接觸的第一電荷傳輸層;第二電極;與第二電極接觸的第二電荷傳輸層;和多個半導體納米晶體,其設置在第一電荷傳輸層和第二電荷傳輸層之間,其中通過配體交換修飾所述多個半導體納米晶體的表面。與尚未具有配體交換表面修飾的光伏器件相比,可測量所述改善的性能。
在某些實施方式中,半導體納米晶體包括硫化鉛。
在某些實施方式中,所述配體可包括硫醇。在某些實施方式中,所述配體可包括胺。在某些實施方式中,所述配體可包括鹵化物。在某些實施方式中,所述配體可包括苯硫酚(BT)、1,2-苯二硫酚、1,3-苯二硫酚、1,4-苯二硫酚、1,2-乙烷二硫醇或3-巰基丙酸。在某些實施方式中,所述配體可包括1,2-乙二胺或硫氰酸銨。在某些實施方式中,所述配體可包括碘化四丁基銨、溴化四丁基銨、氯化四丁基銨或氟化四丁基銨。
另一方面,半導體納米晶體可包括通過配體交換修飾的表面,其中該修飾改善了包括所述半導體納米晶體的光伏器件的性能。
在某些實施方式中,所述配體可包括硫醇。在某些實施方式中,所述配體可包括胺。在某些實施方式中,所述配體可包括鹵化物。在某些實施方式中,所述配體可包括苯硫酚(BT)、1,2-苯二硫酚、1,3-苯二硫酚、1,4-苯二硫酚、1,2-乙烷二硫醇或3-巰基丙酸。在某些實施方式中,所述配體可包括1,2-乙二胺或硫氰酸銨。在某些實施方式中,所述配體可包括碘化四丁基銨、溴化四丁基銨、氯化四丁基銨或氟化四丁基銨。
其它方面、實施方式和特征根據以下描述、附圖和權利要求將是顯然的。
附圖說明
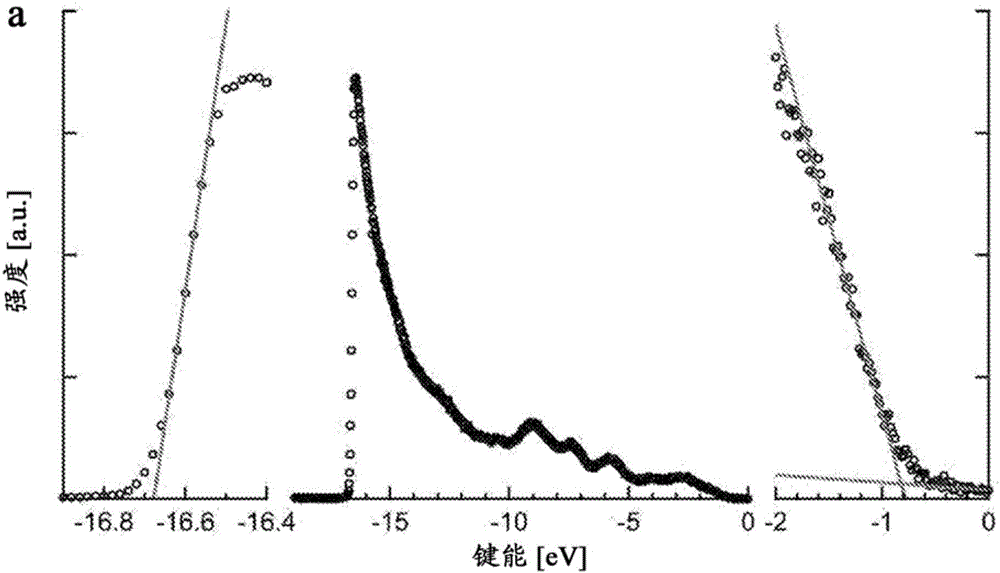
圖1A顯示在金上的100nm厚的1,3-BDT交換的PbS QD膜的完整的紫外光電子光譜;圖1B顯示1,3-BDT交換的PbS QD的第一激子峰(受激峰)的光學吸收光譜(吸收=1-透射-反射);圖1C顯示根據圖1A和1B的光譜以及方程式(1)確定的1,3-BDT交換的PbS QD的能級圖;圖1D顯示配體的化學結構;圖1E顯示通過圖1D中所示的配體交換的PbS QD的完整的能級圖。
圖2A顯示模型化的PbS板層(厚層,slab)的示意圖;圖2B顯示具有不同配體的PbS板層的面均靜電勢;圖2C顯示五種配體各自的配體(填充曲線)和配體-板層體系(未填充曲線)的態密度;圖2D顯示各配體(ΔEvac,黑色箭頭)的真空能量躍遷且分解成界面(ΔEvac,1,紅色箭頭)偶極和固有配體(ΔEvac,2,藍色箭頭)偶極。
圖3A顯示ZnO/PbS np-異質結的光伏性能;圖3B顯示Schottky結構造(architecture)的光伏性能。
圖4A顯示PEDOT:PSS空穴傳輸層的影響;圖4B顯示LiF陰極夾層的影響。
圖5A顯示施主-受主(給體-受體)異質結的器件結構;圖5B顯示施主-受主對的示意性能帶圖;圖5C顯示三個不同尺寸的PbS QD的測量的能級;圖5D顯示與C60配對的不同尺寸的經1,2-BDT處理的QD的電流;圖5E顯示與C60配對的不同尺寸的經1,3-BDT處理的QD的電流;圖5F顯示與C60配對的給定尺寸的經1,2-BDT和1,3-BDT處理的QD的電流;圖5G顯示與PTCBI和C60配對的經1,2-BDT處理的QD的電流。
圖6A顯示量子點的電子性質取決于表面化學;圖6B顯示描繪光伏器件的示意圖。
圖7A顯示在金基底上的100nm厚的苯硫酚配體交換的PbS QD膜的紫外光電子光譜;圖7B顯示在金基底上的100nm厚的1,2-苯二硫酚配體交換的PbS QD膜的紫外光電子光譜;圖7C顯示在金基底上的100nm厚的1,3-苯二硫酚配體交換的PbS QD膜的紫外光電子光譜;圖7D顯示在金基底上的100nm厚的1,4-苯二硫酚配體交換的PbS QD膜的紫外光電子光譜;圖7E顯示在金基底上的100nm厚的1,2-乙烷二硫醇配體交換的PbS QD膜的紫外光電子光譜;圖7F顯示在金基底上的100nm厚的3-巰基丙酸配體交換的PbS QD膜的紫外光電子光譜;圖7G顯示在金基底上的100nm厚的乙二胺配體交換的PbS QD膜的紫外光電子光譜;圖7H顯示在金基底上的100nm厚的硫氰酸銨配體交換的PbS QD膜的紫外光電子光譜;圖7I顯示在金基底上的100nm厚的氟化四丁基銨配體交換的PbS QD膜的紫外光電子光譜;圖7J顯示在金基底上的100nm厚的氯化四丁基銨配體交換的PbS QD膜的紫外光電子光譜;圖7K顯示在金基底上的100nm厚的溴化四丁基銨配體交換的PbS QD膜的紫外光電子光譜;圖7L顯示在金基底上的100nm厚的碘化四丁基銨配體交換的PbS QD膜的紫外光電子光譜。
圖8顯示根據經1,3-BDT處理的PbS QD的UPS光譜確定的能級的時間依賴性(溶液中的第一吸收峰的λ=963nm)。
圖9顯示1,3-BDT交換的PbS QD的UPS結果對QD純化步驟的依賴性(溶液中的第一吸收峰的λ=963nm)。
圖10顯示1,3-BDT交換的PbS QD的光的透射、反射和吸收(溶液中的第一吸收峰的λ=963nm)。
圖11A顯示用苯硫酚進行配體交換的PbS QD的光學吸收光譜;圖11B顯示用1,2-苯二硫酚進行配體交換的PbS QD的光學吸收光譜;圖11C顯示用1,3-苯二硫酚進行配體交換的PbS QD的光學吸收光譜;圖11D顯示用1,4-苯二硫酚進行配體交換的PbS QD的光學吸收光譜;圖11E顯示用1,2-乙烷二硫醇進行配體交換的PbS QD的光學吸收光譜;圖11F顯示用3-巰基丙酸進行配體交換的PbS QD的光學吸收光譜;圖11G顯示用乙二胺進行配體交換的PbS QD的光學吸收光譜;圖11H顯示用硫氰酸銨進行配體交換的PbS QD的光學吸收光譜;圖11I顯示用氟化四丁基銨進行配體交換的PbS QD的光學吸收光譜;圖11J顯示用氯化四丁基銨進行配體交換的PbS QD的光學吸收光譜;圖11K顯示用溴化四丁基銨進行配體交換的PbS QD的光學吸收光譜;圖11L顯示用碘化四丁基銨進行配體交換的PbS QD的光學吸收光譜。
圖12A顯示在溶液中用1,3-BDT進行配體交換的PbS QD的UPS光譜其中第一吸收峰λ=1153nm;圖12B顯示在溶液中用1,2-BDT進行配體交換的PbS QD的UPS光譜其中第一吸收峰λ=1153nm;圖12C顯示在溶液中用1,3-BDT進行配體交換的PbS QD的UPS光譜,其中第一吸收峰λ=905nm;圖12D顯示在溶液中用1,2-BDT進行配體交換的PbS QD的UPS光譜,其中第一吸收峰的λ=905nm;圖12E顯示在溶液中用1,3-BDT進行配體交換的PbS QD的UPS光譜,其中第一吸收峰λ=725nm;圖12F顯示在溶液中用1,2-BDT進行配體交換的PbS QD的UPS光譜,其中第一吸收峰λ=725nm。
圖13顯示1,3-BDT交換的PbS QD的光學吸收光譜。
圖14A顯示模型化的PbS(111)板層的示意圖;圖14B顯示具有不同配體的PbS(111)板層的面均靜電勢。
圖15顯示1,2-BDT和1,3-BDT以多種幾何形狀鍵合(結合,bind)到PbS(100)表面的真空能量躍遷。
圖16A顯示雙面配體鍵合到PbS(100)板層的DFT模擬;圖16B顯示PbS QD FET的過渡曲線(傳遞曲線,transfer curves)。
圖17A顯示使用EDT、1,2-BDT和1,3-BDT配體交換的ZnO/PbS QD NP HJ器件在關閉擾動脈沖之后的ΔV(其中ΔV為由擾動激光脈沖誘導的額外的VOC)的衰減;圖17B顯示NP HJ光伏器件在整個背景光(bias light)強度范圍推導出的復合速率系數krec和誘導的光電壓。
圖18A顯示ZnO/PbS QD NP HJ光伏器件在白光背景下應對擾動光脈沖的瞬態電壓響應;圖18B顯示同一器件不在白光背景的情況下應對相同擾動脈沖的的瞬態電流響應。
圖19顯示阱(trap)密度分布。
圖20A顯示ZnO/PbS QD np異質結光伏器件在用經濾光的氙燈照射下的電流-電壓響應;圖20B顯示同一器件的外量子效率光譜。
具體實施方式
膠體量子點(QD)(也稱為納米晶體(NC))的電子性質極其依賴于QD尺寸和表面化學兩者。量子約束的修飾提供QD帶隙的控制,而配體誘導的表面偶極呈現對電子器件內的QD的絕對能級進行控制的迄今未充分利用的手段。顯示通過紫外光電子光譜法測量的硫化鉛QD的能級在不同化學配體處理之間躍遷高至0.9eV。這些能量躍遷的方向與原子密度泛函理論模擬結果匹配且與配體偶極矩成比例。使用配體修飾的QD膜的光伏器件的性能的趨勢和所測量的能級躍遷一致。這些結果確認了表面化學介導的能級躍遷作為可預測地控制膠體QD膜的電子性質的手段和作為在QD光電子器件的性能優化上的通用可調節參數。
改善光伏器件的性能的方法可包括通過配體交換修飾納米晶體的表面能級。光伏器件可包括這樣的層:其包括具有通過配體交換修飾的表面能的納米晶體。
通過經由配體交換對QD表面化學進行修飾可調整耦合膠體的QD固體的電子性質。改善光伏器件性能的方法可包括通過配體交換修飾納米晶體的表面能級。修飾納米晶體表面的量子約束可控制電子器件內的QD的絕對能級。可使用很多種配體化學性質(chemistry),包括硫醇、伯胺、羧酸、硫氰酸根離子和鹵根離子。
光伏器件可包括第一電極、第二電極、電荷傳輸層和吸收層,該吸收層可包括多個半導體納米晶體。所述納米晶體的表面可通過配體交換進行修飾。修飾納米晶體的表面的量子約束可控制電子器件內的QD的絕對能級且改善所述器件的性能。可使用很多種配體化學性質,包括硫醇、伯胺、羧酸、硫氰酸根離子和鹵根離子
形成納米晶體的半導體可包括第II-VI族化合物、第II-V族化合物、第III-VI族化合物、第III-V族化合物、第IV-VI族化合物、第I-III-VI族化合物、第II-IV-VI族化合物或第II-IV-V族化合物。
當電子和空穴局限在納米晶體上時,發射可在發射波長下發生。該發射具有對應于量子約束的半導體材料的帶隙的頻率。該帶隙是納米晶體的尺寸的函數。具有小直徑的納米晶體可具有介于分子和大塊形式的物質之間的性質。例如,具有小的直徑的基于半導體材料的納米晶體可在整個三維中表現出電子和空穴兩者的量子約束,其導致所述材料的有效帶隙隨著晶粒尺寸的減小而增大。結果,隨著晶粒尺寸減小,納米晶體的光吸收和發射兩者均藍移或或遷移到更高的能量。
來自納米晶體的發射可為窄的Gaussian發射帶,其通過改變納米晶體的尺寸、納米晶體的組成或兩者在遍及紫外、可見或紅外區域的整個光譜進行調整。例如,CdSe可在可見區域中進行調整,且InAs可在紅外區域中進行調整。納米晶體群的窄的尺寸分布可導致窄的光譜范圍內的光的發射。所述群體可為單分散的且可表現出在納米晶體直徑上小于15%、優選小于10%、更優選小于5%的均方根偏差。可觀察到納米晶體的在可見光中發射的不大于約75nm、優選60nm、更優選40nm和最優選30nm的半寬度(FWHM)的窄范圍內的光譜發射。IR-發射的納米晶體可具有不大于150nm或不大于100nm的FWHM。按照發射能量表達,所述發射可具有不大于0.05eV或不大于0.03eV的FWHM。所述發射的寬度隨著納米晶體直徑的分散度的降低而減小。半導體納米晶體可具有例如大于10%、20%、30%、40%、50%、60%、70%或80%的高的發射量子效率。
形成所述納米晶體的半導體可包括第II-VI族化合物、第II-V族化合物、第III-VI族化合物、第III-V族化合物、第IV-VI族化合物、第I-III-VI族化合物、第II-IV-VI族化合物或第II-IV-V族化合物,例如ZnO、ZnS、ZnSe、ZnTe、CdO、CdS、CdSe、CdTe、MgO、MgS、MgSe、MgTe、HgO、HgS、HgSe、HgTe、AlN、AlP、AlAs、AlSb、GaN、GaP、GaAs、GaSb、InN、InP、InAs、InSb、TlN、TlP、TlAs、TlSb、TlSb、PbS、PbSe、PbTe或其混合物。
制備單分散的半導體納米晶體的方法包括被注射到熱的配位溶劑中的有機金屬試劑例如二甲基鎘的熱解。這允許離散的成核且導致宏觀量的納米晶體的受控生長。納米晶體的制備和操縱描述于例如美國專利6,322,901和6,576,291以及美國專利申請No.60/550,314中,其各自通過引用以其整體并入。納米晶體的制造方法為膠體生長工藝。膠體生長通過將M施主和X施主快速注射到熱的配位溶劑中而發生。所述注射產生可以受控方式生長的核而形成納米晶體。可逐漸加熱所述反應混合物以使所述納米晶體生長和退火(韌化,anneal)。樣品中納米晶體的平均尺寸和尺寸分布兩者依賴于生長溫度。保持穩定生長所需的生長溫度隨著平均晶體尺寸的增大而增加。納米晶體是納米晶體群的一員。作為離散成核和受控生長的結果,所獲得的納米晶體群具有窄的、單分散的直徑分布。單分散的直徑分布還可稱為尺寸。成核之后納米晶體在配位溶劑中的受控生長和退火的工藝還可導致均勻的表面延伸(表面衍生,surface derivatization)和規則的芯結構。隨著尺寸分布銳化,可提高所述溫度以保持穩定生長。通過添加更多的M施主或X施主,可縮短生長期。
所述M施主可為無機化合物、有機金屬化合物或金屬單質。M為鎘、鋅、鎂、汞、鋁、鎵、銦或鉈。X施主為能夠與M施主反應而形成具有通式MX的材料的化合物。典型地,X施主為硫屬化物施主或磷屬化物施主,例如膦硫屬化物、雙(甲硅烷基)硫屬化物、雙氧、銨鹽或三(甲硅烷基)磷屬化物。合適的X施主包括雙氧、雙(三甲基甲硅烷基)硒化物((TMS)2Se)、三烷基膦硒化物例如(三正辛基膦)硒化物(TOPSe)或(三正丁基膦)硒化物(TBPSe)、三烷基膦碲化物例如(三正辛基膦)碲化物(TOPTe)或六丙基磷三酰胺碲化物(HPPTTe)、雙(三甲基甲硅烷基)碲化物((TMS)2Te)、雙(三甲基甲硅烷基)硫化物((TMS)2S)、三烷基膦硫化物例如(三正辛基膦)硫化物(TOPS)、銨鹽例如鹵化銨(例如NH4Cl)、三(三甲基甲硅烷基)磷化物((TMS)3P)、三(三甲基甲硅烷基)砷化物((TMS)3As)或三(三甲基甲硅烷基)銻化物((TMS)3Sb)。在某些實施方式中,M施主和X施主可為同一分子內的部分。
配位溶劑可有助于控制納米晶體的生長。配位溶劑為具有給電子孤對(donor lone pair)的化合物,其例如具有可配位到生長的納米晶體表面的孤電子對。配位溶劑可使生長的納米晶體穩定化。典型的配位溶劑包括烷基膦、烷基膦氧化物、烷基膦酸或烷基次膦酸,然而,其它配位溶劑例如吡啶、呋喃和胺也可適宜于納米晶體的制造。合適的配位溶劑的實例包括吡啶、三正辛基膦(TOP)、三正辛基膦氧化物(TOPO)和三羥丙基膦(tHPP)。可使用工業級TOPO。
通過監測粒子的吸收線寬度可估計在所述反應的生長階段期間的尺寸分布。響應于所述粒子的吸收光譜的變化而修正反應溫度容許在生長期間保持尖銳的粒度分布。可在晶體生長期間向成核溶液添加反應物以生長較大的晶體。通過在特定的納米晶體平均直徑時停止生長和選擇合適的半導體材料組成,對于CdSe和CdTe,可在整個300nm-5微米或400nm-800nm的波長范圍內連續地調整納米晶體的發射光譜。納米晶體具有小于的直徑。納米晶體群具有范圍內的平均直徑。
納米晶體可為具有窄尺寸分布的納米晶體群的一員。納米晶體可為球形、棒形、圓盤形或其它形狀。納米晶體可包括半導體材料的芯。納米晶體可包括具有式MX的芯,其中M為鎘、鋅、鎂、汞、鋁、鎵、銦、鉈或其混合物,且X為氧、硫、硒、碲、氮、磷、砷、銻或其混合物。
所述芯可具有在該芯表面上的外敷層(overcoating)。該外敷層可為組成不同于所述芯的組成的的半導體材料。納米晶體的表面上的半導體材料的外敷層可包括第II-VI族化合物、第II-V族化合物、第III-VI族化合物、第III-V族化合物、第IV-VI族化合物、第I-III-VI族化合物、第II-IV-VI族化合物和第II-IV-V族化合物,例如ZnO、ZnS、ZnSe、ZnTe、CdO、CdS、CdSe、CdTe、MgO、MgS、MgSe、MgTe、HgO、HgS、HgSe、HgTe、AlN、AlP、AlAs、AlSb、GaN、GaP、GaAs、GaSb、InN、InP、InAs、InSb、TlN、TlP、TlAs、TlSb、TlSb、PbS、PbSe、PbTe或其混合物。例如,可在CdSe或CdTe納米晶體上生長ZnS、ZnSe或CdS外敷層。外涂敷工藝描述于例如美國專利6,322,901中。通過在外涂敷期間調節反應混合物的溫度和監測所述芯的吸收光譜,可獲得具有高的發射量子效率和窄的尺寸分布的外涂敷的材料。外敷層可為1和10個單層的厚度。
粒度分布可通過用納米晶體的不良溶劑(例如描述于美國專利6,322,901中的甲醇/丁醇)進行尺寸選擇性沉淀而進一步改進。例如,納米晶體可分散于10%丁醇的己烷溶液中。甲醇可逐滴加入到該攪拌著的溶液直至維持乳白色。通過離心將上清液和絮凝物分離,在所述樣品中產生了富含最大晶粒的沉淀物。可重復該步驟直至注意不到光學吸收光譜的進一步尖銳化。可在多種溶劑/非溶劑對(其包括吡啶/己烷和氯仿/甲醇)中進行尺寸選擇性沉淀。尺寸選擇的納米晶體群可具有偏離平均直徑的不超過15%的均方根偏差、優選10%的均方根偏差或更小、和更優選5%的均方根偏差或更小。
納米晶體的外表面可包括源自在生長過程期間使用的配位溶劑的化合物。可通過反復暴露于過量的競爭性配位基團對所述表面進行修飾。例如,包覆的(capped)納米晶體的分散體可用配位的有機化合物例如吡啶進行處理以制造在吡啶、甲醇和芳烴中穩定分散但是無法在脂族溶劑中分散的晶粒。這樣的表面交換工藝可使用能夠配位或鍵合到納米晶體的外表面的任何化合物(例如膦、硫醇、胺和磷酸鹽)進行。可將所述納米晶體暴露于這樣的短鏈聚合物:其對于所述表面呈現親合力且在具有對懸浮液或分散介質具有親合力的部分封端。這樣的親合力改善了所述懸浮液的穩定性且不利于所述納米晶體的絮凝。納米晶體配位化合物描述于例如美國專利No.6,251,303中,其通過引用以其整體并入。
更具體地,配位配體可具有如下式:
其中k為2、3或5,且n為1、2、3、4或5,使得k-n不小于零;X為O、S、S=O、SO2、Se、Se=O、N、N=O、P、P=O、As,或As=O;Y和L各自獨立地為芳基,雜芳基或任選地包括至少一個雙鍵,至少一個三鍵或至少一個雙鍵和一個三鍵的直鏈或支鏈的C2-12烴鏈。所述烴鏈可任選地被一個或多個C1-4烷基、C2-4烯基、C2-4炔基、C1-4烷氧基、羥基、鹵素、氨基、硝基、氰基、C3-5環烷基、3-5員的雜環烷基、芳基、雜芳基、C1-4烷基碳酰氧基、C1-4烷基氧碳酰基、C1-4烷基碳酰基或甲酸基所取代。所述烴鏈還可任選地被-O-、-S-、-N(Ra)-、-N(Ra)-C(O)-O-、-O-C(O)-N(Ra)-、-N(Ra)-C(O)-N(Rb)-、-O-C(O)-O-、-P(Ra)-或-P(O)(Ra)-所隔開(中斷)。Ra和Rb各自獨立地為氫、烷基、烯基、炔基、烷氧基、羥基烷基、羥基或鹵代烷基。
芳基為取代的或未取代的環狀芳香基團。實例包括苯基、芐基、萘基、甲苯基、蒽基、硝基苯基或鹵代苯基。雜芳基為在環中具有一個或多個雜原子的芳基例如呋喃基、吡啶基、吡咯基、菲基。
膠體量子點(QD)擁有一系列獨特的可調的電子性質,這在其作為在溶液加工的光伏器件中的活性材料的應用上已經引起相當大的興趣。參見例如,Kim,J.Y.et al.,Adv.Mater.2013,25,4986–5010,其通過引用以其整體并入。容許再現地控制QD尺寸的合成技術使得能夠制備被強烈約束的硫化鉛(PbS)膠體QD,其中帶隙范圍為0.7-2.1eV,橫跨用于單結和多結光伏器件應用的理想范圍。參見例如,Gao,J.et al.,Nano Lett.2011,11,1002–1008,其通過引用以其整體并入。在通過修飾納米晶體尺寸提供的對于QD帶隙的控制的補充中,也可以通過經由配體交換而修飾QD表面化學性質來調整耦合膠體的QD固體的電子性質。參見例如,Talapin,D.V et al.,Science 2005,310,86–9,其通過引用以其整體并入。對于PbS QD(迄今為止性能最高的QD太陽能電池材料),已經采用很多種化學配體,包括雙配位的脂族和芳族硫醇、伯胺、羧酸、硫氰酸根離子和鹵根離子。對于給定的配體,巖石-鹽-結構PbS納米晶體的不同晶面在空間機會和配體鍵合的親合力上表現另外的差異。參見例如,Ip,A.H et al.,Nat.Nanotechnol.2012,7,577–82;Klem,E.J.D.et al.,Appl.Phys.Lett.2007,90,183113;Pattantyus-Abraham,A.G.et al.,ACS Nano2010,4,3374–80;Fafarman,A.T.et al.,J.Am.Chem.Soc.2011,133,15753–61;Tang,J.et al.,Nat.Mater.2011,10,765–771;Choi,J.J.et al.,J.Am.Chem.Soc.2011,133,3131–8,其各自通過引用以其整體并入。
配體交換可通過改變QD之間的介電環境和隧穿距離而影響載流子遷移率;在不存在其它變化的情況下,遷移率隨著配體長度減小而呈指數增大。參見例如,Liu,Y.et al.,Nano Lett.2010,10,1960–9,其通過引用以其整體并入。合適的配體還可鈍化由QD芯的結構的非周期性和偏離化學計量所引起的在QD表面上的電子阱位點,增加載流子和激子的壽命且提供一定程度的對于耦合的QD膜的摻雜水平和類型的控制。參見例如,Voznyy,O.et al.,ACS Nano2012,6,8448–8455;Oh,S.J.et al.,ACS Nano2013,7,2413–21;Kim,D.et al.,Phys.Rev.Lett.2013,110,196802,其各自通過引用以其整體并入。改變配體的化學鍵合基團的身份和偶極矩也應該改變QD-配體表面偶極的強度,從而改變真空能量,并且進而改變QD價帶頂(VBM)和導帶底(CBM)。QD的表面化學可影響其能級;然而,在OD上用油酸配體(其對于在光伏器件中的使用過于絕緣)或者在QD上用小范圍的其它配體已經實施在PbS QD上進行的大多數能級研究。參見例如,Soreni-Harari,M.et al.,Nano Lett.2008,8,678–84;Timp,B.A.et al.,Surf.Sci.2010,604,1335–1341;Jasieniak,J.et al.,ACS Nano2011,5,5888–902;Munro,A.M.et al.,ACS Appl.Mater.界面s2010,2,863–9;Hyun,B.-R.et al.,ACS Nano2008,2,2206–12;Gao,J.et al.,Nano Lett.2011,11,3263–6;Yuan,M.et al.,Adv.Mater.2013,25,5586–92;Katsiev,K.et al.,Adv.Mater.2013,1–6,其各自通過引用以其整體并入。考慮到其它種類的QD在配體交換之后觀察到的大的能級躍遷,經油酸包覆的PbS QD的能級不可能代表PbS QD太陽能電池中使用的配體交換的膜的能級。
使用紫外光電子光譜法(UPS)測量用十二種不同配體處理的PbS QD的能級躍遷。所測量的價帶頂橫跨0.9eV的范圍。通過將這些不同配體中的五種鍵合到原始的(原生的)PbS板層而誘導的真空能量躍遷的原子學模擬再現了所觀察到的能級修飾的趨勢。這些能級躍遷對光伏性能的影響通過使用1,2-乙烷二硫醇(EDT)、1,2-苯二硫酚(1,2-BDT)和1,3-苯二硫酚(1,3-BDT)配體在器件上進行的研究而確定。即使在這些化學上相似的配體之間,大于0.2eV的VBM躍遷也必須對電子-和空穴提取的觸點(hole-extracting contact)進行依賴于配體的調節以實現最佳性能。最近這些結果已經指導對經證實具有8.55%效率的PbS QD太陽能電池(這類裝置的當前記錄)的設計和認識。參見例如Chuang,C.-H.M.et al.,Nat.Mater.2014,其通過引用以其整體并入。這些發現補充了QD帶隙的可調整性且強調對膠體QD的電子性質進行控制的重要機制。
通過UPS測量的QD能級的配體依賴性
圖1顯示通過UPS測量的依賴于配體的能級。圖1A顯示在金上的100nm厚的1,3-BDT交換的PbS QD膜的完整的紫外光電子光譜。左側和右側的圖片分別示出高截止鍵能(結合能,binding energy)(費米能級)和低截止鍵能(價帶邊緣鍵能)區域的放大圖,其中通過從截止區域線性外推到基線的交叉點確定鍵能。圖1B顯示1,3-BDT交換的PbS QD的第一激子峰的光學吸收光譜(吸收=1–透射–反射)。將E=1.23eV處的峰吸收視作光帶隙。圖1C顯示由A、B中的光譜和方程式(1)確定的1,3-BDT交換的PbS QD的能級圖。在整個多次測量中,區別儀器精度(0.1eV)和標準偏差(0.02eV)。圖1D顯示用在本研究中的配體的化學結構。圖1E顯示用d中所示的配體進行交換的PbS QD的完整的能級圖。該圖中使用的全部PbS QD在含有原生(native)油酸配體的溶液中具有在λ=963nm處的第一激子吸收峰。各數據點表示所有不同樣品中2-4次測量的平均值;陰影條指示一個標準偏差,且為了清楚起見,省去儀器精度的誤差條。
圖1A顯示在沒有暴露于空氣的情況下用1,3-BDT處理的100nm厚的PbS QD膜的代表性UPS光譜。UPS測量占據電子態,且由此提供關于材料的費米能級(低鍵能截止)和VBM(高鍵能截止)的信息。CBM的能量EC可通過將材料的電子傳輸能隙Eg加到VBM而進行估計,其中Eg根據光學帶隙Egopt和受約束的電子和空穴的庫侖穩定能之和而確定,其最初由Brus使用盒內粒子模型(particle-in-a-box model)推導出,使得
其中e是電子的電荷量,ε0是真空(free space)電容率,εQD是QD芯材料的光學介電常數,和R是量子點半徑(通過將溶液中的第一吸收峰與出版公開的粒度曲線(篩分曲線,sizing curve)匹配而確定)。參見例如,Brus,L.,J.Phys.Chem.1986,90,2555–2560,其通過引用以其整體并入。在本研究中使用的PbS QD是高度約束的,其中量子-約束的帶隙0.6–1.1eV大于PbS的本體帶隙。因此,約束的電子和空穴具有高的動能,且相比于靜電介電常數光學介電常數是更合適的選擇。參見例如Dalven,R.,Solid State Phys.1974,28,179–224,其通過引用以其整體并入。
圖1C顯示玻璃上的經1,3-BDT處理的PbS QD的代表性吸收光譜。固態配體交換的QD的第一吸收峰處于對應于Eg=1.32eV的傳輸帶隙的能量Egopt=1.23eV下。圖1C總結了經1,3-BDT處理的PbS QD的根據圖1A和1B中的測量結果確定的VBM、費米能級和CBM的能量。盡管UPS的儀器精度為~0.1eV,但是該測量的標準偏差(這里在所有的4個不同樣品中)小得多,在0.02eV的范圍內。參見例如,Hwang,J.et al.,Mater.Sci.Eng.R Reports 2009,64,1–31,其通過引用以其整體并入。
圖1D顯示在本研究中使用的十二種配體(包括硫醇[苯硫酚(BT),1,2-、1,3-和1,4-苯二硫酚(1,2-BDT、1,3-BDT和1,4-BDT),1,2-乙烷二硫醇(EDT)和3-巰基丙酸(MPA)],伯胺[1,2-乙二胺(EDA)],硫氰酸銨(SCN)和鹵化物[碘化四丁基銨(TBAI),溴化物(TBABr),氯化物(TBACl),和氟化物(TBAF)])的化學結構。圖1E顯示了所測量的單批的用通過降低VBM鍵能分類的這些不同配體交換的PbS QD(溶液中的吸收峰λ=963nm)的能級。
在用TBABr和BT處理的QD之間觀察到VBM中的0.9eV的最大躍遷。甚至在化學上類似的雙配位硫醇中,在用EDT和1,4-BDT處理的PbS QD之間觀察到0.3eV的躍遷。類似地,對于用含胺聚合物的薄層處理的導體,已觀察到大的能級躍遷。這樣的大的躍遷預期對使用PbS QD構造的電子器件的操作具有相當大的影響。這些能級只對于這里研究的PbS QD的特定尺寸且只在無空氣的構造和存儲條件下是特有的;在于空氣中構造的PbS QD膜上進行的UPS測量顯示費米能級和VBM的不同值。還注意到,在本研究中測試的大多數配體引起的VBM比在文獻中報道的油酸包覆的PbS QD的那些顯著更深(油酸過于絕緣而不能用在UPS中,這需要發射表面充分接地以防止在觀察到的能級中發生充電誘導的躍遷)。該結果強調了在盡可能接近于在操作器件存在的環境的化學環境中對于QD進行能級測量,既要考慮固態配體環境又要考慮空氣、真空和溶劑的暴露歷史的重要性。
通過密度泛函理論的配體結合模擬
第一定律密度泛函理論(DFT)計算提供對于通過UPS測量的帶能能躍遷的根源的洞察。DFT計算廣泛用于模擬無機材料和有機分子之間的的界面處的能級躍遷。參見例如,Zhou,Y.et al.,Science2012,336,327–32;Heimel,G.et al.,Nano Lett.2007,7,932–40;Yang,S.et al.,Nano Lett.2012,12,383–8,其各自通過引用以其整體并入。圖2顯示PbS板層的配體誘導的能量躍遷的DFT計算。圖2A顯示模型化的PbS板層的示意圖。所述板層的左側被吸附的配體(此處顯示1,2-BDT作為實例)所鈍化,而右側被合適的贗(擬)氫原子所鈍化以保證電荷平衡。單配位(BT,碘化物)和雙配位(1,2-、1,3-和1,4-BDT)配體用在這里,其中配體密度設定在每個表面Pb原子一個結合原子(因此,雙配位的配體具有單配位配體一半的面密度)。圖2B顯示具有不同配體的PbS板層的面均靜電勢。將真空區域中遠離未鈍化的PbS板層左側的電勢設定為零。圖2C顯示所考慮的五種配體各自的配體(填充曲線)和配體-板層體系(未填充曲線)的態密度。各鈍化的PbS板層上方的真空水平設定為零。豎直的虛線標明價帶和導帶的邊緣能量。圖2D顯示各配體的真空能量躍遷(ΔEvac,黑色箭頭)且分解為界面(ΔEvac,1,紅色箭頭)偶極和固有配體(ΔEvac,2,藍色箭頭)偶極。
如圖2A中所示,PbS QD的表面近似成半無限的PbS(100)板層,因為對于PbS QD,(100)和(111)面可占優勢(對于鍵合到富Pb的(111)面,獲得DFT結果中的類似趨勢)。參見例如,Cho,K.-S.et al.,J.Am.Chem.Soc.2005,127,7140–7;Bealing,C.R.et al.,ACS Nano2012,6,2118–27,其各自通過引用以其整體并入。以作為半無限的準二維板層的QD表面建模比以整個三維QD/配體體系建模在計算上高效得多,且電子或空穴在轉移期間在整個QD/配體界面上所遇到的靜電環境在兩種情形中應該是類似的。對于InAs QD,在其它地方已經觀察到配體誘導的躍遷的數量級對QD尺寸的依賴性小,但是不同配體的能級趨勢的方向預期與QD尺寸無關。如圖2A中所示,板層的一側被配體鈍化,且為了電荷平衡,一側由贗氫原子鈍化。當板層的兩側均由配體鈍化時獲得類似結果。
以上使用的五種配體(BT、1,2-BDT、1,3-BDT、1,4-BDT和碘化物)通過DFT進行模擬,其中配體覆蓋率保持在每表面Pb原子一個配體鍵合基團不變(因此,BT和碘化物具有兩倍的苯二硫酚密度)。這里所測試的配體可高效地取代初始的油酸配體,但是或許由于(100)面和(111)面之間的鍵合親合力改變的結果,一些油酸可能保持與QD表面鍵合。參見例如,Luther,J.M.et al.,ACS Nano2008,2,271–80;Tang,J.et al.,ACS Nano2010,4,869–78;Koh,W.-K.et al.,Nano Lett.2011,11,4764–7;Law,M.et al.,J.Am.Chem.Soc.2008,130,5974–85;Yaacobi-Gross,N.et al,Combining Ligand-Induced Quantum-Confined Stark Effect with Type II Heterojunction Bilayer結構in CdTe.2012;Yaacobi-Gross,N.et al.,Nat.Mater.2011,10,974–9,其各自通過引用以其整體并入。為了便于在不同配體之間比較且簡化DFT模擬,假設油酸完全交換。
圖2B顯示結合到PbS(100)板層的這五種配體的面均靜電勢。觀察到相比于未鈍化的PbS板層的大的真空能級的躍遷(ΔEvac)。圖2C顯示對于所述五種配體各自的配體(填充曲線)和配體-板層體系(未填充曲線)的電子態密度。PbS帶隙在配體吸附時保持相對不變,而VBM和CBM躍遷與圖2B中觀察到的ΔEvac匹配,表明了該帶邊躍遷本質上是靜電的。盡管通過DFT高估了躍遷的數量級,但是與通過UPS和DFT觀察到的帶邊躍遷的方向和排序存在優異的一致性。
配體吸附時的QD能級躍遷可概念化為兩個偶極貢獻之和:來自QD的表面原子和配體的鍵合基團之間形成的偶極的貢獻(這里稱為μ1)和來自配體本身的固有偶極矩的貢獻(μ2)。對于這里所研究的路易斯堿性配體,μ1從帶負電的配體指向帶正電的鉛原子;μ2取決于配體的化學結構和鍵合取向。總偶極的z-分量(μtotal,z)可表示成μtotal,z=μ1,z+μ2,z,通過Helmholtz方程式將其與ΔEvac關聯:
其中A為配體的表面積,和εr為配體層的介電常數。參見例如Zehner,R.W.et al.,Langmuir1999,15,1121–1127;Alloway,D.M.et al.,J.Phys.Chem.B2003,107,11690–11699;Boer,B.de et al.,Adv.Mater.2005,17,621–625,其各自通過引用以其整體并入。
圖2D顯示各配體的ΔEvac和由ΔEvac分解成的反向ΔEvac,1和ΔEvac,2項。配體-固有的ΔEvac,2項遵循簡單靜電學預測的趨勢:碘化物不具有固有偶極,而硫醇的ΔEvac,2隨著C-S鍵之間的角度減小而增大。界面ΔEvac,1項對于小型的(緊湊的)碘化物和BT配體是大的,且對于空間體積更大的配體則變小。ΔEvac的趨勢是由配體偶極矩而不是界面偶極的影響所支配,因為ΔEvac隨著|ΔEvac,2|的減小而單調地增大。與表面-配體偶極矩反向的固有偶極矩的缺失是鹵化物配體的一般特征,且解釋了在圖1E中對于該類配體所觀察到的大的鍵能躍遷。多個配體類別與圖1E中觀察到的趨勢的高度一致支持了UPS在可靠測量QD能級上的應用和本文呈現的能級躍遷的直觀描述。
依賴于配體的光伏性能
為了確定這些QD能級躍遷對于光伏器件的重要性,將用EDT、1,2-BDT和1,3-BDT交換的PbS QD摻入到ZnO/PbS QD np異質結、Schottky結和施主-受主異質結光伏器件中。這些配體迄今已經在PbS QD光電子器件中充分研究,且使用相同的、可再現的配體交換步驟,從而它們提供用于對比的理想平臺。參見例如Leschkies,K.S.et al.,ACS Nano2009,3,3638–48;Johnston,K.W.et al.,Appl.Phys.Lett.2008,92,151115;Zhao,N.et al.,ACS Nano2010,4,3743–52;Brown,P.R.et al.,Nano Lett.2011,11,2955–61;Jeong,K.S.et al.,ACS Nano2012,6,89–99,其各自通過引用以其整體并入。這里所研究的光伏器件的所有QD膜的制備在和以上描述的UPS研究的膜的制備均在相同的無空氣和無水的條件下進行。
圖3顯示依賴于構造的光伏性能。對于EDT-、1,2-BDT-和1,3-BDT交換的PbS QD(溶液中的第一吸收峰的λ=905nm),圖3A中的ZnO/PbS np-異質結和圖3B的Schottky結構造,在黑暗(虛線)中和在100mW cm-2的AM1.5照射(實線)下測量電流-電壓特性。
圖3顯示用EDT-、1,2-BDT和1,3-BDT交換的PbS QD(溶液中的第一吸收峰的λ=905nm)膜制作的氧化銦錫(ITO)/ZnO/PbS QD/MoO3/Au np-異質結(np-HJ)光伏器件(圖3A)和ITO/PEDOT:PSS/PbS QD/LiF/Al Schottky結(SJ)光伏器件(圖3B)的黑暗和光照J-V特性。兩者的總體性能趨勢是顯然的。第一,EDT處理在np-HJ和SJ構造兩者中導致比1,2-BDT和1,3-BDT處理低的開路電壓(VOC)。第二,1,2-BDT和1,3-BDT交換的QD的相對性能在np-HJ和SJ構造之間反轉:1,3-BDT交換對于np-HJ構造導致最佳的性能,而1,2-BDT交換對于SJ構造導致最佳的性能。阱分布、載流子遷移率和復合率的直接對比可解釋這兩種趨勢的第一種,但是無法解釋第二種。盡管EDT處理在所研究的三種配體中導致最高的載流子遷移率,但是EDT交換的PbS QD膜的高復合率和高阱密度導致比BDT交換的膜低的VOC和功率轉換效率(ηP)。然而,這些性質的變化沒有解釋1,2-BDT和1,3-BDT交換的QD的依賴于構造的性能。np-HJ和SJ構造之間的QD膜能量環境的差異暗示了,兩種不同配體處理之間的PbS QD的能級的躍遷可解釋所述性能的差異。
憑借聚焦在SJ構造上,可通過修飾提取電子和空穴的觸點以受控的方式調整界面能。簡單的ITO/PbS/陰極結構的修飾為,在ITO和PbS QD之間插入PEDOT:PSS層作為提取電子的層。參見例如Ma,W.et al.,2011,8140–8147;Choi,J.J.et al.,Nano Lett.2009,9,3749–55,其各自通過引用以其整體并入。由于其深的(大的,deep)功函(對于PEDOT:PSS,EF=5.0eV,相對于ITO的4.7eV)的結果,EDOT:PSS可有助于將空穴注入到具有深的最高占據分子軌道(HOMO)的有機半導體中和將空穴從該有機半導體中提取出。參見例如Koch,N.et al.,Appl.Phys.Lett.2003,82,70;Milliron,D.J.et al.,J.Appl.Phys.2000,87,572,其各自通過引用以其整體并入。
圖4顯示配體誘導的Schottky光伏性能的變化。圖4描繪使用1,2-BDT(紅色跡線)和1,3-BDT(藍色跡線)的Schottky結光伏器件的電流-電壓特性,顯示PEDOT:PSS空穴傳輸層(圖4A)和LiF陰極夾層(圖4B)的影響。在各情形中,所述夾層以和圖1的結果一致的方式顯著改善所述兩種配體中僅一種的性能。在圖4A中,觀察到,包括PEDOT:PSS空穴提取層導致用1,3-BDT交換的PbS QD制作的SJ光伏器件的ηP改善3.2-倍,而對1,2-BDT交換的SJ光伏器件具有可忽略的影響。該觀察結果和由圖1E中報道的能級所預期的相配:經1,3-BDT處理的QD由于其較深的VBM比經1,2-BDT處理的QD更多地受益于高功函的PEDOT:PSS空穴傳輸層。
類似地,LiF薄層可插入有機LED和太陽能電池中的陰極和電子傳輸層之間,其中顯示電子注入和提取的效率得以增加。參見例如Shaheen,S.E.et al.,J.Appl.Phys.1998,84,2324;Tang,J.et al.,Adv.Mater.2010,22,1398–402,其各自通過引用以其整體并入。該效果可歸因于作為由LiF誘導的強界面偶極的結果的有效陰極功函下降,從而導致電子注入的勢壘高度降低。參見例如Brabec,C.J.et al.,Appl.Phys.Lett.2002,80,1288,其通過引用以其整體并入。在這里描述的PbS QD SJ構造中,陰極功函的降低應該對具有較淺的(較小的,shallower)VBM和CBM的PbS QD通過增強界面處的Schottky結和增大電子提取的驅動力帶來更大的益處。確實,在圖4B中觀察到,LiF的插入對于經1,2-BDT處理的QD導致2.2-倍的ηP改善,同時對于經1,3-BDT處理的QD的ηP具有可忽略的影響,這和圖1E中所報導的經1,2-BDT處理的PbS QD的較淺的能級一致。
施主-受主異質結(DA-HJ)是SJ構造的可選方案,該SJ構造直接依賴于用于分離電荷載流子的在D-A界面處的帶偏移(而不是可對表面阱和其它復雜因素敏感的Schottky勢壘的形成),從而提供其中可更直接地探測界面能級校直(對準,align)的構造。參見例如Luther,J.M.et al.,Nano Lett.2008,8,3488–92,其通過引用以其整體并入。圖5顯示配體-誘導的和QD-尺寸-誘導的DA-HJ光伏性能的變化。圖5A顯示施主-受主異質結的器件結構,和圖5B顯示施主-受主對的示意性能帶圖,顯示對于光電流提取有利的導帶偏移ΔECB。圖5C顯示三種不同尺寸的具有C60和來自文獻的PTCBI的LUMO能量的PbS QD的所測量的能級。導帶能EC對應于傳輸能隙;為了清楚起見,這里省略了光學能隙。QD能級的通過UPS確定的實驗不確定度為0.1eV;由文獻中的逆光電子光譜確定的有機材料的LUMO的不確定度為0.5eV。圖5D-5G顯示DA-HJ光伏器件的暗電流(短劃線曲線)、亮電流(實曲線)和光電流(點曲線),比較了與C60配對的不同尺寸的經1,2-BDT處理的QD的圖5D、與C60配對的不同尺寸的經1,3-BDT處理的QD的圖5E、與C60配對的給定尺寸的經1,2-BDT和1,3-BDT處理的QD的圖5F、以及與PTCBI和C60配對的經1,2-BDT處理的QD的圖5G。
圖5A顯現其中PbS QD充當電子施主并且巴克敏斯特富勒烯(C60)或3,4,9,10苝四羧酸二苯并咪唑(PTCBI)充當電子受主的DA-HJ器件構造。圖5B顯現DA-HJ光伏器件的能級結構的略圖。DA-HJ的一個重要的設計標準在于,通過ΔECB=ECBMD–ECBMA給出的ΔECB必須是正的且足夠大以容許來自施主的CBM的光生電子轉移到受主的最低未占據分子軌道(LUMO)。大的正ΔECB還應該阻礙不希望的光生電子從受主逆轉移(反向轉移)到施主。因此,通過修飾ΔECB和觀察所得的DA-HJ光伏器件的性能,可推測PbS QD的能級躍遷且將其與通過UPS確定的那些進行比較。
圖5C顯示三種不同尺寸的PbS QD(溶液中的第一吸收峰的λ=725nm、905nm和1153nm)在用1,2-BDT或1,3-BDT的配體交換之后的測量的能級。正如之前在文獻中已經注意到的,發現了CBM隨QD尺寸的變化比VBM大。C60和PTCBI的LUMO取自文獻中報道的逆光電子光譜學測量結果,分別為4.0±0.5eV和3.6±0.5eV。圖5D-5G顯示將這三種不同尺寸的經1,2-BDT-和1,3-BDT交換的PbS QD與C60和PTCBI配對的DA-HJ光伏器件的暗電流、亮電流和光電流J-V響應(分別為J暗電流、J亮電流和J光電流,其中J光電流=J亮電流-J暗電流)。
隨著經1,2-BDT交換的QD的帶隙減小,準費米能級分裂降低的容量導致較小的VOC,但是在較長波長下的吸收增加導致較高的JSC(圖5D)。對于1,3-BDT交換的QD,觀察到相同的趨勢(圖5E),但是1,3-BDT的二極管性質相當大地偏離理想行為。隨著帶隙減小且CBM移向更深的能量,J亮電流在更小的電壓下與J暗電流交叉,且前部偏壓J亮電流的“彎折(kink)”變得更顯著,這與J光電流極性的反轉對應,因為短路電流(負極性)被增大的光電導率(正極性)所容納。如果使施主-受主界面處的ΔECB變得不那么正(對應于施主CBM的加深),則預期到前部偏壓中光電導率增大,因為降低了電子從受主轉移到施主的勢壘。
當比較PTCBI和C60作為受主的性能以及經1,2-BDT交換的QD和經1,3-BDT交換的QD作為施主的性能時,在圖5F、g中觀察到類似趨勢。PTCBI的LUMO在能量上比C60的LUMO淺~0.4eV,從而導致ΔECB減小;相應地,使用PTCBI的DA-HJ證實來自以較低電壓J亮電流-J暗電流交叉點形式的光電導逆電子轉移的較大貢獻。類似地,根據圖5C,測量的經1,3-BDT交換的1.1eV PbS QD的CBM比相同尺寸的1,2-BDT交換的PbS QD的CBM深0.2eV,這還導致ΔECB減小;如此,1,3-BDT的J亮電流表明較強的前部偏壓“彎折”和與J暗電流的較低的電壓交叉點。
總的來說,圖5D-5G顯示用1,3-BDT代替1,2-BDT(根據這里報道的能級測量結果,其導致0.1-0.2eV更深的CBM)誘導DA-HJ光伏性能改變,該變化與通過減小QD的帶隙或通過取代C60的PTCBI(其兩者均可導致ΔECB減小)誘導的那些在性質上類似。此觀察為這里所報道的能級測量方法和這些能級在描述PbS QD光電子器件的性能上的應用提供支持。
膠體QD的帶能可通過配體交換進行修飾,從而導致PbS QD的高至0.9eV的能級躍遷。能級位置在不同配體之間的趨勢通過原子學模型進行確認,顯示出觀察到的躍遷由來自QD-配體界面偶極和配體分子本身的固有偶極矩兩者的貢獻所致。這些能級躍遷導致光伏器件操作的可預測的變化而且提供對于QD光伏器件的最佳配體選擇和器件構造的指導。這些可指導使用級聯的能級構造設計當前記錄效率(current-record-efficiency)的PbS QD光伏器件。作為量子約束受控的帶隙修飾的補充,這些結果驗證了配體誘導的帶能躍遷作為可預測地控制膠體QD的電子性質的手段和在優化QD光電子器件上關鍵的可調參數。
光伏器件可包括第一電極;與第一電極接觸的第一電荷傳輸層;第二電極;與第二電極接觸的第二電荷傳輸層;和設置在第一電荷傳輸層和第二電荷傳輸層之間的多個半導體納米晶體,其中所述多個半導體納米晶體的表面通過通過配體交換修飾。
第一電荷傳輸層可包括第一無機材料。該第一無機材料可為無定形的或多晶的。該第一無機材料可為無機半導體。該無機半導體可包括金屬硫屬化物。該金屬硫屬化物可為混合的金屬硫屬化物。該金屬硫屬化物可包括氧化鋅、氧化鈦、氧化鈮、硫化鋅、氧化銦錫,或其混合物。
第二電荷傳輸層可包括第二無機材料。該第二無機材料可為無定形的或多晶的。該第二無機材料可為無機半導體。該無機半導體可包括金屬硫屬化物。該金屬硫屬化物可為混合的金屬硫屬化物。該金屬硫屬化物可包括氧化鋅、氧化鈦、氧化鈮、硫化鋅、氧化銦錫、氧化鉬,或其混合物。
光伏器件可具有例如圖6B中所示的結構,其中第一電極2、第一層3與電極2接觸,第二層4與所述層3接觸,且第二電極5與第二層4接觸。第一層3可為空穴傳輸層且第二層4可為電子傳輸層。至少一個層可為非聚合物的。所述層可包括無機材料。所述結構的電極之一與基底1接觸。各電極可接觸電源以在整個所述結構中提供電壓。當在整個器件上施加合適的極性和大小的電壓時光電流可通過吸收層而產生。第一層3可包括多個半導體納米晶體,例如基本上單分散的納米晶體群。納米晶體的表面可通過配體交換修飾。納米晶體的表面可通過配體交換修飾。
替代地,在空穴傳輸層和電子傳輸層之間可包括獨立的吸收層(圖6B中未示出)。該獨立的吸收層可包括多個納米晶體。所述納米晶體的表面可通過配體交換修飾。包括納米晶體的層可為納米晶體的單層或納米晶體的多層。在一些情況下,包括納米晶體的層可為不完整的層,即包含缺少材料的區域的層,使得鄰近所述納米晶體層的層可為部分接觸的。所述納米晶體和至少一個電極具有足以將電荷載流子從納米晶體轉移到第一電極或第二電極的帶隙偏移。所述電荷載流子可為空穴或電子。所述電極轉移電荷載流子的能力允許光誘導的電流以便于光檢測的方式流動。
實施例
PbS QD的合成和膜的制備。
全部合成、制作和測試均在無氧氣和無水條件下進行,除非另有聲明。油酸包覆的PbS QD經由標準的文獻方法合成,且將其通過三次在丙酮和1-丁醇的混合物中沉淀和離心隨后在己烷中再懸浮而純化。參見例如Chang,L.-Y.et al.,Nano Lett.2013,13,994–9,其通過引用以其整體并入。在最后一輪純化之后,將QD以25mg mL-1的濃度溶解在辛烷中。將所有固體QD膜通過順序旋轉涂膜(spin-casting)而制備。對于每層,將~15μL的QD溶液通過0.02μm的過濾器(Anotop)分配到12.5mm x 12.5mm基底上并以1500rpm旋轉涂膜15s。然后將大致200μL的配體溶液通過0.1μm過濾器(PTFE)分配到所述基底上,容許其靜置30s,和將其旋轉干燥。然后,以用于配體交換的溶劑將基底淹沒,且將其旋轉干燥三次以除去未結合的配體,以及重復整個過程;每次完整的循環導致沉積~20nm的QD。用在該研究中的如下配體濃度和溶劑代表了文獻中充分表征的配體交換條件:在乙腈(ACN)中1.7mM的苯硫酚以及1,2-、1,3-、1,4-苯二硫酚;在ACN中1.7mM的1,2-乙烷二硫醇;在甲醇(MeOH)中115mM的3-巰基丙酸;在MeOH中1M的乙二胺;在MeOH中30mM的硫氰酸銨;以及在MeOH中30mM的氟化四丁基銨、氯化物、溴化物和碘化物。參見例如Koleilat,G.I.et al.,ACS Nano2008,2,833–40;Talgorn,E.et al.,Nat.Nanotechnol.2011,6,733–739;Zhitomirsky,D.et al.,N-Type Colloidal-Quantum-Dot Solids for Photovoltaics.2012,其各自通過引用以其整體并入。全部化學品以可獲得的最高純度購自Sigma Aldrich。
紫外光電子光譜。
使用具有10-10毫巴的基礎壓力的Omicron超高真空系統收集UPS光譜。用于UPS測量的基底為通過熱蒸鍍涂覆有Cr(10nm)/Au(100nm)陽極的、且在沒有進一步表面處理的情況下儲存于無空氣條件中的12.5mm x 12.5mm的載玻片。用如上述,進行多種配體處理的PbS QD的五個順序旋轉涂膜循環,產生~100nm的QD膜厚。使用碳帶進行從鋼樣品板至Cr/Au陽極的電接觸。使用裝載鎖定的(load-locked)轉移系統在沒有暴露于空氣的情況下將UPS樣品從惰性氣氛的手套箱(<1ppm O2)輸送到UPS系統。在UPS測量期間,通過來自氦放電燈的He(I)發射線提供21.22eV下的照射,且腔室壓力增大到10-7毫巴。所述樣品加偏壓-5.0V以保證精確確定低動能截止(cutoff),且在相對于法線0°收集電子發射。單次動能掃描以<45秒完成以使充電最小化。由截止區域的線性外推與基線的線性外推的交叉點確定截止能量。
密度泛函理論的計算。
使用具有用于交換和關聯函數的Perdew-Burke-Ernzerhof(PBE)的概括的梯度近似的Vienna Ab從頭算模擬軟件包(Vienna Abinitio Simulation Packages)(VASP)進行DFT計算。參見例如Kresse,G.et al.,Comput.Mater.Sci.1996,6,15–50;Perdew,J.et al.,Phys.Rev.Lett.1996,77,3865–3868,其各自通過引用以其整體并入。采納投影綴加波來描述芯電子。近期工作已經表明,配體覆蓋密度的差異可顯著影響電子性質;13這里的覆蓋率保持在每個表面鉛原子一個鍵合原子不變。在大量的集中分析之后使用400eV的截止能量和5x5x1的Monkhorst-Pack k-點取樣。使用的大的真空間隔以防止板層內的相互作用。各(100)PbS板層由8個單層組成,且選擇具有5/3的分數電荷和1/3e的贗氫原子來鈍化底層上的表面Pb和S原子。使用共軛梯度法使配體和PbS板層的上面五層完全弛豫直至該結構滿足以下弛豫標準:(i)兩個連續的離子步驟之間的能量差小于10-4eV,和(ii)作用在各原子上的最大Hellmann-Feynman力小于包括偶極關聯以除去在鄰近的超級單元(supercell)之間的欺騙性靜電相互作用。
光伏器件的制作。
將光伏器件沉積到如下經ITO涂覆的玻璃基底(薄膜器件)上:在微-90皂液、去離子水、丙酮和異丙醇中已超聲清洗,隨后使用臭氧等離子體進行處理。陽極-陰極重疊限定的器件面積為1.24mm2。通過射頻濺射沉積氧化鋅(質粒)。氧化鉬(99.9995%)、氟化鋰(99.99%)、巴克敏斯特富勒烯(C60)、3,4,9,10苝四羧酸二苯并咪唑(PTCBI)、浴銅靈(BCP)、鋁、銀和金在10-6托的基礎壓力下通過熱蒸鍍以進行沉積。通過旋轉涂膜將聚乙烯二氧噻吩-聚苯乙烯磺酸鹽(PEDOT:PSS,導電級,Sigma-Aldrich)在空氣中以4000rpm沉積60s,然后在氮氣氣氛手套箱內在150℃下退火30分鐘。經ITO涂覆的玻璃基底在12mM(3-巰基丙基)三甲氧基硅烷(3-MPTMS)的甲苯溶液中浸漬過夜以增大QD對基底的粘附力,然后在異丙醇中超聲1分鐘以除去未鍵合的3-MPTMS。Np-異質結(np-HJ)光伏器件采用構造ITO/ZnO(50nm)/PbS QD(160nm)/MoO3(10nm)/Au(100nm)。Schottky結光伏器件采用構造ITO/PEDOT:PSS/PbS QD(160nm)/LiF(0.7nm)/Al(100nm),除非另外指出。施主-受主光伏器件采用構造ITO/PEDOT:PSS/PbS QD(160nm)/(C60或PTCBI)(40nm)/BCP(10nm)/Ag(100nm)。
電學表征。
使用Keithley 6487皮可安培計記錄光伏器件的電流密度-電壓(J-V)曲線,且通過裝備有AM1.5G濾光器的氙燈(Newport 96000)提供100mW cm2的照射。
QD-配體絡合物的紫外光電子光譜和吸收光譜
圖7顯示100nm厚的配體交換的PbS QD膜在金基底上的紫外光電子光譜。圖7顯示100nm厚的配體交換的PbS QD膜在金基底上的紫外光電子光譜:A,苯硫酚;B,1,2-苯二硫酚;C,1,3-苯二硫酚;D,1,4-苯二硫酚;E,1,2-乙烷二硫醇;F,3-巰基丙酸;G,乙二胺;H,硫氰酸銨;I,氟化四丁基銨;J,氯化四丁基銨;K,溴化四丁基銨;L,碘化四丁基銨。展示每個樣品在五分鐘收集的五次連續掃描。紅線表示第一次掃描的擬合(標記為“1分鐘”)。費米能級根據二次電子截止區域的線性外推與x-軸的截距而確定。價帶鍵能由一次電子截止區域線性外推與基線的線性外推的截距而確定。
圖7顯示用于確定圖1中所示的配體交換的PbS QD(溶液中的第一吸收峰的λ=963nm)的能級的代表性紫外光電子光譜。顯示了各配體的以一分鐘間隔收集的五次順序掃描。對于平衡中的未變樣品,所述五次掃描應該相同;如果樣品由于不充分的電接觸隨時間而充電或如果樣品的化學結構由于與21.22eV的光子束的相互作用改變,則光譜隨時間可改變。觀察到,硫醇的光譜在五分鐘的間隔內非常恒定;鹵化物配體的光譜經歷小的朝向較淺功函的遷移(~0.1eV)。對于本工作中報道的能級,只使用來自給定樣品的第一次掃描(這里標記為“1分鐘”)以使充電和樣品損傷最小化(有機材料或混雜的有機-無機材料的UPS研究中的慣例),且對由此確定的2-4個單獨樣品的能級取平均以產生圖1E呈現的最終值。參見例如Greiner,M.T.et al.,Nat.Mater.2012,11,76–81,其通過引用以其整體并入。
圖8顯示1,3-BDT交換的PbS QD在九次順序掃描內的價帶頂(VBM)的能量和費米能級。價帶能量和費米能級在此時間間隔內均沒有經歷躍遷。圖8顯示根據經1,3-BDT處理的PbS QD(溶液中的第一吸收峰的λ=963nm)的UPS光譜確定的能級的時間依賴性。在圖8中,這里的誤差棒表示實驗精確度。沒有觀察到實驗上顯著的能級差異。
鑒于UPS對于表面化學性質的變化的高靈敏性,核實在樣品制備和樣品歷史上的不希望的差異沒有影響測量的能級是重要的。圖1中采用的全部量子點均來自單個合成批次,因此QD尺寸上的差異應該是最小的。QD的表面化學性質和配體覆蓋率可潛在地受到純化步驟和在離心和重新懸浮之前非溶劑將QD“撞出(沖洗出,crashed out)”溶液所用強度的影響。圖9顯示在如下三次不同的撞出步驟之后由用1,3-BDT交換的PbS QD的UPS光譜確定的能級:在沉淀步驟中采用加入最小量的丙酮的“軟”撞出、使用大大過量的丙酮的“硬”撞出、和使用乙醇代替丙酮的撞出。觀察到,這些不同撞出步驟均導致相同的能級值,表明圖1中報道的差異是由于配體交換所用的配體而不是在QD合成或純化中的差異引起的。還值得注意的是,樣品在UPS測試期間暴露于超高真空(UHV)的時長僅為~1小時,與在太陽能電池制作的觸點蒸發期間將QD樣品暴露于高真空的時長相當。圖9顯示1,3-BDT交換的PbS QD(溶液中的第一吸收峰的λ=963nm)的UPS結果對QD純化步驟的依賴性。在圖9中,這里的誤差棒表示實驗精確度。沒有觀察到實驗上顯著的能級差異。
圖10顯示在短暫地暴露于空氣期間測量的1,3-BDT交換的PbS QD(溶液中的第一吸收峰的λ=963nm)的代表性光透射、反射和吸收光譜,其中A%=100%-T%-R%。如在方法部分中所描述的,將100nm厚的QD膜(使用旋轉涂膜且配體交換的QD的5個順序層)沉積到用3-巰基丙基三甲氧基硅烷處理的玻璃上。以相對于法線8°的入射角測量反射。沒有考慮散射;散射的存在引起所計算的吸收曲線的偏移,但是不影響根據吸收光譜的第一激子峰確定的帶隙值。圖10顯示1,3-BDT交換的PbS QD(溶液中的第一吸收峰的λ=963nm)的光透射、反射和吸收。
圖11顯示配體交換的PbS QD的光學吸收光譜。在圖11中,將硫化鉛QDs(溶液中的第一吸收峰的λ=963nm)用如下進行交換:A,苯硫酚;B,1,2-苯二硫酚;C,1,3-苯二硫酚;D,1,4-苯二硫酚;E,1,2-乙烷二硫醇;F,3-巰基丙酸;G,乙二胺;H,硫氰酸銨;I,氟化四丁基銨;J,氯化四丁基銨;K,溴化四丁基銨;L,碘化四丁基銨。
圖11顯示用圖1中所示的十二種配體的每一種交換的、在短暫地暴露于空氣期間收集的PbS QD的光學吸收光譜(溶液中的第一吸收峰的λ=963nm)。光學帶隙Egopt由所示數據的11點取樣積分(矩形窗,boxcar)平均值的第一激子吸收特征峰的能量確定。傳輸帶隙Eg根據如下方程式確定:
其中e是電子的電荷量,ε0為真空電容率,εQD為QD芯材料的光學介電常數(ε∞PbS=17.2),和R為量子點半徑。參見例如Brus,L.J.Phys.Chem.1986,90,2555–2560;Dalven,R.Solid State Phys.1974,28,179–224,其各自通過引用以其整體并入。通過將溶液中的第一吸收峰與篩分曲線的冪律外推相匹配來確定QD半徑。QD上的電荷和其在QD和配體母體(matrix)之間的整個介電邊界上誘導的圖像電荷之間的相互作用引起的另外的穩定作用采取如下形式:
其中εmatrix為配體殼層的光學介電常數;之前的研究已經表明該項在配體交換的QD體系中是可忽略的,因而其在這里被忽略不計。參見例如Jasieniak,J.et al.,ACS Nano2011,5,5888–902,其通過引用以其整體并入。圖13顯示圖5中采用的旋涂到玻璃上且用1,3-BDT配體交換的三種尺寸的PbS QD的光學吸收光譜。如上所述,光學帶隙根據第一激子吸收特征峰的能量(對于其中只觀察到肩峰的最高帶隙點,根據肩峰區域中吸收分布的第二衍生物的最小值確定光學帶隙)而確定,且傳輸帶隙使用Jasieniak中的篩分曲線根據方程式(1)其確定。參見Jasieniak,J.et al.,ACS Nano 2011,5,5888–902,其通過引用以其整體并入。圖13顯示圖5使用中的的1,3-BDT交換的PbS QD的光學吸收光譜,用D、QD直徑和λsoln(溶液中的第一吸收峰)標記。為了清楚起見將光譜垂直偏移。
圖12顯示不同尺寸的PbS QD的UPS光譜。在圖12中,將硫化鉛QD以~100nm厚的層(5個順序旋轉涂膜周期)沉積到金基底上。樣品由如下組成:具有溶液中第一吸收峰λ=1153nm的通過A,1,3-BDT和B,1,2-BDT交換的QD;具有溶液中第一吸收峰λ=905nm的通過C,1,3-BDT和D,1,2-BDT交換的QD;以及具有溶液中第一吸收峰λ=725nm的通過E,1,3-BDT和F,1,2-BDT交換的QD。
圖12顯示所收集的用在圖5中的經1,2-BDT-和1,3-BDT交換的PbS QD的UPS光譜。如同在圖7中,顯示了五次順序掃描,表明光譜隨時間沒有相當大的變化;在各情形中只使用第一次掃描的擬合來確定能級。
在富Pb的PbS(111)表面上的配體鍵合模擬
可模擬將配體鍵合到PbS(111)表面的原子學DFT以補充鍵合到PbS(100)表面上的討論,且(100)面和(111)面兩者對于PbS QD均可占主導。參見例如Choi,J.J.et al.,J.Am.Chem.Soc.2011,133,3131–8;Cho,K.-S.et al.,J.Am.Chem.Soc.2005,127,7140–7;Bealing,C.R.et al.,ACS Nano2012,6,2118–27,其各自通過引用以其整體并入。可模擬將BT、1,2-BDT、1,3-BDT和碘化物配體鍵合到富鉛的PbS(111)平面;發現1,4-BDT在PbS(111)表面上的鍵合是不穩定的。圖14A顯示模型化的PbS(111)板層的示意圖。具有5/3e分數電荷量的贗氫原子使右邊的鉛原子鈍化,且配體(1,2-BDT在圖10A中作為實例示出)使左邊的鉛原子鈍化。圖14B顯示具有不同配體的PbS(111)板層的面均靜電勢曲線圖。不同配體表現出不同的真空能級躍遷(ΔEvac):對于碘化物、1,3-BDT、1,2-BDT和BT,真空能量躍遷分別為ΔEvac=+2.58eV、+0.08eV、-0.03eV和-0.39eV。這種鍵合到富鉛的PbS(111)表面的真空能量的趨勢方向和圖2中觀察到的鍵合到PbS(100)表面的趨勢方向相同。
圖14顯示鍵合到PbS(111)表面的配體誘導的能級躍遷的DFT計算。圖14A顯示模型化的PbS(111)板層的示意圖。板層的左側通過所述配體(1,2-BDT在這里作為實例示出)鈍化,且右側通過合適的贗氫原子鈍化以保證電荷平衡。圖14B顯示具有不同配體的PbS(111)板層的面均靜電勢。遠離未鈍化PbS板層的左邊的真空區域中的電勢設定為零。這里觀察到的能級躍遷的趨勢方向和圖2中觀察到的趨勢匹配。
單配位和雙配位的配體在PbS(100)表面上的鍵合模擬
用在這里的雙配位的硫醇配體(1,2-BDT、1,3-BDT、1,4,-BDT和EDT)可以多種構型鍵合到PbS(100)表面。在單配位連接中,給定配體的兩個硫醇基團之一鍵合到單個QD作為硫醇鹽;第二硫醇被質子化且未鍵合到QD或鍵合到鄰近的QD。在雙配位連接中,硫醇基團的兩個均鍵合到單個QD;對于PbS(100)面,這些硫醇將鍵合到最近的鄰Pb原子(具有的沿<110>方向的間隔)或鍵合到最近的鄰Pb原子(具有的沿<100>方向的間隔)的下一個。
研究了1,2-BDT和1,3-BDT在PbS(100)表面上的單個單配位鍵合模式和兩個雙配位鍵合模式。圖15呈現兩種配體的這三種鍵合模式各自的DFT計算的結果。對于所考慮的這三種鍵合模式的每一種,1,3-BDT的ΔEvac比1,2-BDT的高;該ΔEvac躍遷導致經1,3-BDT處理的PbS QD的比經1,2-BDT處理的PbS QD深的價帶,再現了圖1中通過UPS觀察到的和在圖4-5中通過光伏測量觀察到的趨勢。圖15顯示1,2-BDT和1,3-BDT以多種幾何構型鍵合到PbS(100)表面的真空能量躍遷。在圖15中,各構型的能級躍遷ΔEvac分解成來自界面偶極(紅色;正向貢獻)和固有配體偶極(藍色;負向貢獻)的分量。這里的ΔE對應于給定鍵合模式(單配位模式;以<110>配體取向的雙配位模式;或以<100>配體取向的雙配位模式)的1,2-BDT和1,3-BDT之間的真空能量躍遷差異。
雙側配體鈍化的PbS板層的模擬。
所述模擬將QD表面作為半無限的擬二維板層進行處理,其中配體鍵合到板層的一側,且電荷平衡的贗氫原子鍵合到另一側。采用該近似以提高計算效率和使不同配體之間的對比變容易。實踐中,預期到三維QD的全部表面被配體所鈍化。為了驗證該單側鍵合模式且將該模擬移向更接近物理現實的一步,相比于未鈍化的PbS(100)板層,在兩側均被碘化物配體鈍化的PbS(100)板層上進行DFT模擬。圖16顯示這兩個體系的面均靜電勢曲線圖。對于各情形選擇任意的y-軸偏移,使得靜電勢在PbS板層的中央處校直,且未鈍化的板層外側的真空能量為零。這里觀察到的雙側碘化物鈍化的真空能量躍遷ΔEvac=1.88eV與所觀察到的單側碘化物鈍化的真空能量躍遷ΔEvac=1.90eV幾乎一致。圖16A顯示雙側配體鍵合到PbS(100)板層的DFT模擬。在圖16中,未鈍化的PbS板層外側的真空能量設定為零,且選擇碘化物鈍化的PbS板層的任意偏移,使得靜電勢在板層的中央處校直。觀察到這里所觀察的雙側碘化物鈍化的真空能量躍遷(ΔEvac=1.88eV)和所觀察的單側碘化物鈍化的真空能量躍遷(ΔEvac=1.90eV)之間的幾乎一致。圖16B顯示PbS QD FET的轉換曲線。上部的圖提供所述器件結構的示意性橫截面。
配體交換的PbS QD的場效應遷移率、復合率和態密度
如圖3中所示,通過EDT、1,2-BDT和1,3-BDT交換的PbS QD與ZnO/PbS np異質結(NP HJ)和Schottky結(SJ)構造表現不同。如果阱密度和載流子遷移率、密度和復合率是用這些配體處理的PbS QD之間的差異的唯一來源,則將預期對于兩種構造在這三種配體處理之間具有相同的相對性能;如果能級是差異的唯一來源,則不同配體的性能的等級次序將不變或在兩種構造之間反轉。相反,EDT在兩種構造中表現出最差的性能;1,3-BDT在NP HJ構造中優于1,2-BDT,而1,2-BDT在SJ構造中優于1,3-BDT。正如所預期的,配體交換在阱和載流子兩者的性質和QD的能級上引起變化。這里,通過組合場效應遷移率、復合和阱密度的測量,可量化所測試的三種配體之間的阱和載流子性質的差異,表明它們可解釋EDT的差的性能但是不能解釋1,2-BDT和1,3-BDT性能上的變化。
場效應晶體管制作和測試。場效應晶體管(FET)器件采用底接觸頂柵(bottom-contact top-gate)的幾何構型。簡單來說,源極和漏極分別由被熱蒸鍍到玻璃上和通過剝離(liftoff)(薄膜裝置,Thin Film Device)圖案化的具有L=10um和W=12mm的長度(L)和寬度(W)的Cr(3nm)/Au(40nm)相互交錯的陣列組成。如在方法部分中所描述的,基底用3-MPTMS進行預處理,且通過順序旋轉涂膜沉積配體交換的PbS QD單層。將柵極絕緣層950PMMA A4抗蝕劑(Microchem)以1000rpm旋轉涂膜~470nm的厚度。借助熱蒸鍍通過蔭罩沉積頂接觸鋁柵極。使用Agilent 4156C半導體參數分析儀進行FET測量。以<400ms進行獨立的電流-電壓掃掠(sweep)以使偏壓應力(stress)最小化。使用Solartron 1260阻抗分析儀測量不具有QD的器件的絕緣的柵極絕緣層的電容。
瞬態-VOC測量。在ZnO/PbS NP HJ器件上進行瞬態-VOC測量。通過來自氙燈的穿過液體光導管和中性密度濾光器到達活性區域的白光對器件加偏壓。通過可調的中性密度濾光器由在11Hz處斬切的(chop)且共聚焦到活性區域上的λ=635nm激光提供低強度的擾動。調節激光的強度使得電壓擾動ΔV小于通過白光背景產生的VOC的5%。使用高阻抗輸入的Tektronix3054B示波器根據擾動脈沖的下降邊(后沿,trailing edge)來收集瞬態-電壓曲線。
態密度測量。進行態密度測量。如同在上述的瞬態-VOC測量中,通過氙燈提供強度可調的白色背景光。將通過使用Agilent 33220A信號發生器調制λ=635nm激光的輸出而產生的300ns持續時間的激光脈沖共聚焦到所述活性區域上。使用中性密度濾光器調制白光背景強度和激光脈沖強度使得電壓擾動ΔV小于由白光背景產生的VOC的5%。使用設定為1MΩ輸入阻抗的Tektronix 3054B示波器測量瞬態響應:使用ADA400A高阻抗探針收集電壓跡線,和使用DHPCA-100前置放大器收集電流跡線。
配體交換的PbS QD膜的載流子遷移率由在線性方案中在5V的絕對漏極-源極偏移(|VDS|)下收集的FET轉換特性取得,如圖16中所示。電子和空穴的遷移率分別通過將IDS-VG曲線的線性區域擬合到如下方程式由n-通道和p-通道響應而確定:
其中IDS為漏極-源極電流,VG為柵極偏壓,L和W分別為所述通道的長度(10μm)和寬度(12mm),和Ci為絕緣的柵極絕緣層的電容(其測量為5.7nF cm-2)。所觀察到的滯后現象由QD通道內的偏壓-應力所致;該現象在別處嚴格地驗證過。參見例如Osedach,T.P.et al.,ACS Nano2012,6,3121–7;Ip,A.H.et al.,Nat.Nanotechnol.2012,7,577–82,其各自通過引用以其整體并入。遷移率由起始于VG=0且掃描至較高的絕對柵極偏壓的轉換曲線的分支獲取。所獲取的各配體處理的電子遷移率μe和空穴遷移率μh為:對于EDT,μe=5x10-3cm2V-1s-1和μh 6x10-4cm2V-1s-1;對于1,2-BDT(沒有觀察到n-通道打開),μh=1x10-5cm2V-1s-1;對于1,3-BDT,μe=2x10-4cm2V-1s-1和μh=9x10-6cm2V-1s-1。因此,經EDT處理的PbS QD膜表現出比經1,2-BDT-或1,3-BDT處理的膜顯著更高的載流子遷移率。
在配體之間不存在其它差異下,較高的載流子遷移率本應導致使用EDT交換的PbS QD的光伏器件具有較高的性能;反而,經1,2-BDT和1,3-BDT交換的PbS QD在NP HJ和SJ構造中均表現出較高的性能。使用經1,2-BDT-、1,3-BDT和EDT交換的QDNP HJ光伏器件的瞬態電流和電壓分析表明EDT交換的QD的較高遷移率比其較高的載流子復合率和阱密度重要。
圖17顯示瞬態-VOC分析。圖17A顯示使用EDT、1,2-BDT和1,3-BDT配體交換的ZnO/PbS QD NP HJ器件在關掉擾動脈沖之后的ΔV(其中ΔV為由擾動激光脈沖誘導的額外的VOC)衰減。圖17B顯示NP HJ光伏器件在背景光強度和誘導的光電壓范圍上獲取的復合速率系數krec。對于各誘導的VOC,調節擾動源的強度使得ΔVmax小于VOC的5%。EDT和1,3-BDT的復合速率系數對應于單指數衰減動力學的擬合;顯示了1,2-BDT的單指數擬合和雙指數擬合兩者的復合速率系數。
圖17A顯示通過白光照射加偏壓VOC=0.41V的ZnO/PbS QD NP HJ光伏器件在關掉擾動照射脈沖之后即刻的瞬態電壓響應。EDT處理導致比1,2-BDT和1,3-BDT處理快得多的電壓衰減。EDT和1,3-BDT的該衰減符合單指數動力學,而1,2-BDT的衰減符合雙指數動力學,從而使得能夠計算復合率常數krec。圖17B顯示經EDT-、1,2-BDT-和1,3-BDT交換的PbS QD在NP HJ光伏器件中在背景光強度和誘導電壓范圍下的獲取的krec值。觀察到,在給定的電壓和光生電荷密度下,EDT導致比苯二硫酚的任一種大的復合速率系數。較高的復合率轉變為較高的暗電流和較低的VOC,從而解釋了所觀察到的使用EDT的光伏器件的VOC相對于使用1,2-BDT和1,3-BDT的那些低。
對于EDT的較高復合率背后的機理的洞察通過分析電子阱狀態在帶隙內的密度分布而獲得。所有測量在ZnO/PbS QD NP HJ器件上進行。將所述器件保持于在給定的VOC和對應的在白光背景的情況下通過照射而分裂的準費米能級。短的擾動激光脈沖(300ns的持續時間)產生過大的載流子密度Δn,從而導致光電壓ΔV增大。對于給定的載流子密度Δn,誘導的光電壓ΔV取決于對應于特定VOC的準費米能級下的態密度:對于給定的Δn,較大的態密度將導致較小的ΔV,因為準費米能級僅增大到足以填充Δn狀態。因此,通過對VOC隨Δn/ΔV的變化進行繪圖,可確定電子狀態在帶隙內的密度分布(即阱狀態的密度)。
圖18A顯示1,3-BDT交換的PbS QD NP HJ器件在給定的白光背景下在用給定強度的擾動激光脈沖激發之后的電壓響應。所誘導的電壓脈沖的高度給出ΔV。為了確定該擾動強度的Δn,關掉白光背景且測量所述器件對于單獨的擾動脈沖的瞬態電流響應,如圖18B中所示。該完整的電流分布的積分提供該擾動強度的ΔQ=AeΔn(A為器件面積;e為電子的電荷量)。圖18A顯示ZnO/PbS QD NP HJ光伏器件在白光背景下對于擾動光脈沖的瞬態電壓響應。電壓響應繪制在左軸,且擾動脈沖的分布繪制在右軸。電壓峰在基線上方的高度等于ΔV。圖18B顯示該器件在沒有白光背景的情況下對于相同的擾動脈沖的瞬態電流響應。電流響應繪制在左軸,且積分的電荷量繪制在右軸。積分的電荷量的最右值得到用于計算態密度分布的ΔQ=AeΔn的值。
通過改變背景光的強度(和相應的誘導的VOC)且在各強度下測量ΔV和Δn,獲得Δn/ΔV隨著準費米能級分裂變化的完整曲線。調節擾動激光脈沖的強度以在各背景光強度下保持ΔV小于VOC的5%。圖19顯示使用EDT-、1,2-BDT和1,3-BDT交換的QD的ZnO/PbS QD NP HJ光伏器件的經計算的態密度分布。EDT的態密度比1,2-BDT或1,3-BDT的態密度增加快得多,對應于經EDT處理的PbS QD的帶隙內的阱狀態的較大的密度。應注意,該方法沒有規定所測量的態密度是對應于電子狀體還是空穴狀態;因為電子和空穴準費米能級在較高的VOC值下實現較大的分離,所以電子阱和空穴阱兩者均有助于所述響應。圖19顯示使用以EDT、1,2-BDT或1,3-BDT配體交換的ZnO/PbS QD NP HJ光伏器件在Δn/ΔV方面測量的阱密度分布。
因此,EDT交換的PbS QD的較高的復合率和阱密度比其較高的載流子遷移率重要,其解釋了經EDT處理的PbS QD在NP HJ構造和SJ構造兩者中產生比經1,2-BDT和1,3-BDT處理的PbS QD低的VOC的原因。然而,這里所描述的遷移率、復合率和阱密度上的差異無法解釋NP HJ和SJ構造中的1,2-BDT和1,3-BDT之間的性能差異。這些發現連同本文所報道的數據一起0強.調了在比較QD光電子器件的不同配體處理中考慮載流子傳輸性質和偶極誘導的能級躍遷兩者的變化的重要性。
太陽能電池光譜失配
以未填滿照射幾何形式使來自氙燈(Thermo Oriel 66921)的斬切白光通過單色器進入單芯光纖中并到達有源器件而進行外量子效率測量;器件電流使用鎖定放大器(Stanford Research Systems SR830)測量。
圖20顯示使用經1,3-BDT處理的QD的ZnO/PbS QD NP HJ器件的測量的J-V和外量子效率(EQE)響應。對EQE光譜和AM1.5G太陽能光譜的乘積積分得到預期的在15.0mA cm-2太陽光照射下的JSC;與16.2mA cm-2的測量JSC的對比得到0.92光譜失配。圖20A顯示ZnO/PbS QD np異質結光伏器件在用經濾光的氙燈的照射下的電流-電壓響應。圖20B顯示該器件的外量子效率光譜。
其它實施方式在以下權利要求的范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!