一種β?羰基磷酸酯的制備方法與流程
本發明涉及有機物合成制備技術領域,具體而言,涉及一種β-羰基磷酸酯的制備方法。
背景技術:
β-羰基磷酸酯是一類非常重要的有機化合物和反應中間體,在有機合成及藥物化學中占據重要地位。β-羰基磷酸酯本身具有廣泛的生物學活性和很強的金屬絡合能力,在醫藥、生命科學、功能材料等許多方面有著廣泛的應用。因此,β-羰基磷酸酯的合成工作一直備受科研工作者的關注。
一個多世紀以來,科研工作者對β-羰基磷酸酯的合成方法的研究從未間斷過,合成β-羰基磷酸酯的方法更是層出不窮。目前報道的合成β-羰基磷酸酯的方法主要有:(1)經典的米凱利斯-阿爾布佐夫重排反應(michaelis-arbuzovrearrangement);(2)有機磷酸酯的酰化;(3)炔基磷酸酯的水合作用;(4)烯烴的氧化磷酰化作用。但這些已報道的方法都有一定的局限性,如低原子經濟、原料不易得、反應步驟繁瑣、反應條件苛刻、產率低、需要大量有機金屬參與等。
在前列腺素20余種藥物合成中,很多種都采用β-羰基磷酸酯作為一個重要的中間體,通過著名的horner-wadsworth-emmons(hwe)反應來合成α,β-不飽和羰基化合物,進而通過進一步轉化形成前列腺素類藥物的一個側鏈。而這類β-羰基磷酸酯主要是通過磷酸酯類化合物的酰化制備。目前,普遍使用的制備途徑是甲基磷酸酯在-78℃低溫及無水無氧條件下,在丁基鋰強堿的作用下,與羧酸衍生物反應得到β-羰基磷酸酯。該制備途徑,反應條件苛刻,在放大反應時由于引入易燃的丁基鋰,需要額外注意操作的作安全性。2009年johny.l.chung課題組提出了一種較為簡便、快捷(反應時間僅需15min)的β-羰基磷酸酯的制備方法。該方法以烷基磷酸酯和羧酸衍生物為原料,通過酰化反應得到目標化合物。但是,該方法仍然存在一些缺點,例如需要大量lda參與反應,且反應過中仍需要控制反應溫度為0℃以下,而lda的制備最終還得需要低溫下利用丁基鋰進行。因此,仍然不能解決β-羰基磷酸酯制備條件苛刻和操作存在潛在安全隱患的問題。
有鑒于此,特提出本發明。
技術實現要素:
本發明的第一目的在于提供一種β-羰基磷酸酯的制備方法,以環氧化合物與亞磷酸三烷基酯進行區域選擇性的開環反應生成β-羥基磷酸酯中間體,隨后氧化得到β-羰基磷酸酯,所述的制備方法,具有反應條件溫和、底物適應范圍廣、不需要低溫及無水無氧等苛刻的反應條件,以及操作簡便安全等優點,適合大規模生產。
為了實現本發明的上述目的,特采用以下技術方案:
一種β-羰基磷酸酯的制備方法,包括以下步驟:
以環氧化合物與亞磷酸三烷基酯進行區域選擇性的開環反應,生成β-羥基磷酸酯的含羥基的中間體,然后將中間體的羥基氧化得到β-羰基磷酸酯;
所述環氧化合物的結構式為:
優選的,所述結構式中,r選自芳基、烷基、烷基醚、芳基醚中的一種。
優選的,所述環氧化合物的制備方法,包括以下步驟:
以端基烯烴作原料,將端基烯烴溶于二氯甲烷中,加入氧化劑進行氧化反應,得到環氧化合物;
或者;
將環氧氯丙烷中的-cl替代為r基,形成環氧化合物。
優選的,所述氧化劑選自間氯過氧苯甲酸或過氧硫酸氫鉀;
更優選的,所述氧化劑與所述端基烯烴的摩爾比為(1~1.2):1;
更優選的,所述氧化反應的時間為2~5小時,溫度為0~30℃;
更進一步優選的,所述氧化反應結束后,加入亞硫酸鈉水溶液攪拌除去多余氧化劑,經二氯甲烷萃取、飽和碳酸氫鈉水溶液洗滌,減壓濃縮除去萃取溶劑得到環氧化合物。
優選的,所述區域選擇性的開環反應,使用zncl2溶液體系。
優選的,當使用所述zncl2溶液體系時,具體包括以下步驟:
環氧化合物和亞磷酸烷基酯溶于溶劑中,加入zncl2催化劑,加熱攪拌,采用tlc或hplc監控反應進程,得到的粗產物用于下一步反應;
更優選的,所述溶劑選自乙醚、正己烷、苯和甲苯中的一種,更進一步優選的溶劑為甲苯;
更優選的,每毫摩爾所述環氧化合物使用3~8毫升溶劑,更進一步優選的為5毫升;
更優選的,所述加熱攪拌的溫度為40~80℃,更進一步優選的為60℃。
優選的,所述環氧化合物、所述亞磷酸烷基酯與所述zncl2催化劑的摩爾比為1:(2.0~4.0):(0.3~1.0),更進一步優選的摩爾比為1:3:0.5。
優選的,所述羥基氧化的氧化劑,選自dess-martin氧化劑、pdc體系氧化劑、swern體系氧化劑、瓊斯試劑中的一種;
更優選的,所述氧化劑為瓊斯試劑。
優選的,所述瓊斯試劑氧化的方法,具體包括以下步驟:
丙酮作溶劑,在所述中間體中加入瓊斯試劑,室溫下攪拌,采用tlc或hplc監控反應進程,反應完畢后,處理得到粗產物經純化得到純品β-羰基磷酸酯;
更優選的,每毫摩爾所述環氧化合物使用3~8毫升溶劑,更進一步優選為5毫升。
優選的,所述環氧化合物與所述氧化劑摩爾比為1:(1.5~5.0),更優選的摩爾比為1:2。
與現有技術相比,本發明的有益效果為:
(1)本申請所提供的β-羰基磷酸酯的制備方法,以環氧化合物與亞磷酸三烷基酯進行區域選擇性的開環反應生成β-羥基磷酸酯中間體,隨后氧化得到β-羰基磷酸酯,該方法反應條件溫和,操作簡便,無需無水溶劑及氧氣的保護,更加適合規模化生產。
(2)本申請所提供的β-羰基磷酸酯的制備方法所合成的β-羰基磷酸酯,可以作為多種前列腺素藥物合成的重要中間體,例如曲沃前列腺素、比馬前列腺素及他氟前列腺素等。
附圖說明
為了更清楚地說明本發明具體實施方式或現有技術中的技術方案,下面將對具體實施方式或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖是本發明的一些實施方式,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。
圖1為本發明實施例1所得到的產物的hplc圖;
圖2為本發明實施例2所得到的產物的hplc圖;
圖3為本發明實施例3所得到的產物的hplc圖。
具體實施方式
下面將結合附圖和具體實施方式對本發明的技術方案進行清楚、完整地描述,但是本領域技術人員將會理解,下列所描述的實施例是本發明一部分實施例,而不是全部的實施例,僅用于說明本發明,而不應視為限制本發明的范圍。基于本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。實施例中未注明具體條件者,按照常規條件或制造商建議的條件進行。所用試劑或儀器未注明生產廠商者,均為可以通過市售購買獲得的常規產品。
一種β-羰基磷酸酯的制備方法,包括以下步驟:
以環氧化合物與亞磷酸三烷基酯進行區域選擇性的開環反應,生成β-羥基磷酸酯的含羥基的中間體,然后將中間體的羥基氧化得到β-羰基磷酸酯;
所述環氧化合物的結構式為:
在本申請中,可以采用以下路線進行合成:
以環氧化合物與亞磷酸三烷基酯進行區域選擇性的開環反應生成β-羥基磷酸酯中間體,隨后氧化得到β-羰基磷酸酯,該方法反應條件溫和,操作簡便,無需無水溶劑及氧氣的保護,更加適合規模化生產。該制備方法所合成的β-羰基磷酸酯,可以作為多種前列腺素藥物合成的重要中間體,例如曲沃前列腺素、比馬前列腺素及他氟前列腺素等。
優選的,所述結構式中,r選自芳基、烷基、烷基醚、芳基醚中的一種。
芳基在有機化學中是指任何從簡單芳香環衍生出的官能團或取代基。烷基是一類僅含有碳、氫兩種原子的鏈狀有機官能團,如甲基、乙基、丙基、異丙基、正丁基、異丁基、仲丁基、叔丁基。
在本申請中,如果環氧化合物需要合成,可以采用以下兩種方法進行合成。
優選的,所述環氧化合物的制備方法,包括以下步驟:
以端基烯烴作原料,將端基烯烴溶于二氯甲烷中,加入氧化劑進行氧化反應,得到環氧化合物;
或者;
將環氧氯丙烷中的-cl替代為r基,形成環氧化合物。
當環氧化合物需要制備時,可以采用以上兩種合成路線進行合成。
優選的,所述氧化劑選自間氯過氧苯甲酸或過氧硫酸氫鉀;
更優選的,所述氧化劑與所述端基烯烴的摩爾比為(1~1.2):1;
更優選的,所述氧化反應的時間為2~5小時,溫度為0~30℃;
更進一步優選的,所述氧化反應結束后,加入亞硫酸鈉水溶液攪拌除去多余氧化劑,經二氯甲烷萃取、飽和碳酸氫鈉水溶液洗滌,減壓濃縮除去萃取溶劑得到環氧化合物。
優選的,所述區域選擇性的開環反應,使用zncl2溶液體系。
優選的,當使用所述zncl2溶液體系時,具體包括以下步驟:
環氧化合物和亞磷酸烷基酯溶于溶劑中,加入zncl2催化劑,加熱攪拌,采用tlc或hplc監控反應進程,得到的粗產物用于下一步反應。
tlc為薄層色譜分析,hplc為高效液相色譜分析。
更優選的,所述溶劑選自乙醚、正己烷、苯和甲苯中的一種,更進一步優選的溶劑為甲苯;
更優選的,每毫摩爾所述環氧化合物使用3~8毫升溶劑,更進一步優選的為5毫升;
更優選的,所述加熱攪拌的溫度為40~80℃,更進一步優選的為60℃。
優選的,所述環氧化合物、所述亞磷酸烷基酯與所述zncl2催化劑的摩爾比為1:(2.0~4.0):(0.3~1.0),更進一步優選的摩爾比為1:3:0.5。
優選的,所述羥基氧化的氧化劑,選自dess-martin氧化劑、pdc體系氧化劑、swern體系氧化劑、瓊斯試劑中的一種;
更優選的,所述氧化劑為瓊斯試劑。
優選的,所述瓊斯試劑氧化的方法,具體包括以下步驟:
丙酮作溶劑,在所述中間體中加入瓊斯試劑,室溫下攪拌,采用tlc或hplc監控反應進程,反應完畢后,處理得到粗產物經純化得到純品β-羰基磷酸酯;
更優選的,每毫摩爾所述環氧化合物使用3~8毫升溶劑,更進一步優選為5毫升。
優選的,所述環氧化合物與所述氧化劑摩爾比為1:(1.5~5.0),更優選的摩爾比為1:2。
實施例1
本實施例采用以下合成路線進行制備:
具體操作包括以下步驟:
在500ml三口瓶中,室溫下(大約25℃)將51.84g3-三氟甲基苯酚(320mmol,1.0eq.)懸濁在160ml水中。然后加入氫氧化鈉水溶液[由7.68gnaoh(192mmol,0.6eq.)溶于40ml水中制成],反應體系變澄清。在攪拌40分鐘后,加入75.6ml環氧氯丙烷(960mmol,3eq.),隨后反應體系在30~35℃下攪拌約16小時,液相檢測原料反應完全后,反應體系冷卻至室溫,轉移到分液漏斗中,分出下層中間體產品于500ml燒瓶中,加入160ml水,然后加入氫氧化鈉水溶液[由12.8gnaoh(320mmol,1eq)]溶于40ml水中制成)。反應體系在室溫下(大約25℃)攪拌10小時,液相檢測反應完全。
分離下層產品,加入100ml二氯甲烷形成溶液,加入50ml水進行洗滌,然后無水硫酸鈉干燥,濃縮得到66.27g產物,產率為95%,核磁顯示已較純,可以直接使用于下一步。
1hnmr[400mhz,cdcl3]δppm:7.41(t,j=8.4and7.6hz,1h),7.24(d,j=8.0hz,1h),7.15(s,1h),7.11(d,j=8.4hz,1h),4.31(dd,j=2.8and10.8hz,1h),4.00(dd,j=5.6and10.8hz,1h),3.38(d,j=2.0hz,1h),2.94(dd,j=4.4and8.8hz,1h),2.79(dd,j=3.2and4.0hz,1h)
在100ml燒瓶中,分別加入2.18g上一步產品(10mmol,1eq.),3.72g亞磷酸三甲酯(30mmol,3eq.)和50ml甲苯。在攪拌下,加入1.34g氯化鋅(10mmol,1eq.),然后反應體系加熱到60℃在氬氣下攪拌約10小時,液相檢測氯離子開環副產物生成(約10%及約5%原料)剩余。冷卻反應體系至室溫,加入50ml甲基叔丁基醚溶解,然后分別用飽和碳酸氫鈉水溶液(30ml)和飽和食鹽水(20ml)洗滌。無水硫酸鈉干燥,濃縮除掉多余亞磷酸三甲酯得粗產品,不經分離純化直接使用于下一步。
1hnmr[400mhz,cdcl3]δppm:7.41(t,j=8.0hz,1h),7.23(d,j=7.6hz,1h),7.14(s,1h),7.10(d,j=8.4hz,1h),4.43(m,1h),4.16(m,2h),3.85(s,3h),3.80(s,3h),3.73(br,s,1h,-oh),2.24(m,2h)。
100ml燒瓶中,2.85g上一步粗產品(按理論值計算,應為3.30g,10mmol,1.0eq.)溶解在50ml丙酮中,然后冰水浴下6分鐘內滴加3mljones試劑(8n-cro3:5.33g溶于4.4ml濃硫酸中,然后加入15.6ml水稀釋得到20ml溶液)(24mmol,2.4eq.)在反應體系攪拌4h后,液相檢測原料有少量剩余。然后又補加1mljones試劑,攪拌2小時后,加入5ml異丙醇,攪拌1小時后,過濾,二氯甲烷洗滌,減壓蒸餾除去二氯甲烷、丙酮及異丙醇溶劑。加入30ml水,經二氯甲烷萃取(30ml*2),然后有機層用水溶液(20ml)洗滌。無水硫酸鈉干燥,濃縮得到粗產品(2.35g)。考慮到雜質極性相對很小,而產品極性卻非常大,因而純化相對非常容易。最終經短柱層析(開始用ea/pe=1:2沖/洗,小極性雜質很快除掉,隨后用ea/meoh=9:1沖洗給出產品)純化得到1.70g產品,產率為55%。
1hnmr[400mhz,cdcl3]δppm:7.42(t,j=8.0hz,1h),7.26(d,j=6.8hz,1h),7.14(s,1h),7.11(d,j=8.4hz,1h),4.81(s,2h),3.75(s,3h),3.68(s,3h),3.30(d,m,j=23.2hz,2h)。
實施例2
本實施例采用以下合成路線進行制備:
在1000ml三口瓶中,室溫下(大約25℃)將60g苯酚(638mmol,1.0eq.)懸濁在320ml水中。然后加入氫氧化鈉水溶液[由13.4gnaoh(335mmol,0.6eq.)溶于80ml水中制成],反應體系變澄清。在攪拌40分鐘后,加入178.4g環氧氯丙烷(1920mmol,3eq.),隨后反應體系在30~35℃下攪拌約16小時,液相檢測原料反應完全后,反應體系冷卻至室溫,轉移到分液漏斗中,分出下層中間體產品于1000ml燒瓶中,加入320ml水,然后加入氫氧化鈉水溶液[由26gnaoh(640mmol,1eq)]溶于80ml水中制成)。反應體系在室溫下(大約25℃)攪拌10小時,液相檢測反應完全。分離下層液體產品96g,產率為100%,核磁顯示已較純,可以直接使用于下一步。
在1000ml燒瓶中,分別加入90g上一步產品(600mmol,1eq.)、223g亞磷酸三甲酯(1800mmol,3eq.)和500ml甲苯。反應體系加熱到65~70℃,在攪拌下,分批少量加入82g氯化鋅(600mmol,1eq.),然后加熱攪拌約6小時,原料反應完畢。冷卻后,減壓蒸餾將甲苯等溶劑濃縮至油狀物,加入二氯甲烷(500ml)和稀鹽酸(10%,300ml),分層,水相用二氯甲烷(300ml)反萃,合并二氯甲烷萃取液并濃縮得到油狀產物粗品,不經分離純化直接使用于下一步。
上一步粗產品(按理論值計算,600mmol,1.0eq.)溶解在1000ml丙酮中,然后冰水浴下2小時內滴加240mljones試劑(8n-cro3:64g溶于52.8ml濃硫酸中,然后加入187.2ml水稀釋得到240ml溶液)(640mmol,1.1eq.),在反應體系攪拌5h后,檢測原料反應完全,加入40ml異丙醇,攪拌3小時后,過濾,丙酮洗滌,減壓蒸餾溶劑。加入200ml水,經二氯甲烷萃取(200ml*2),然后有機相濃縮得到粗產品,最終通過柱層析純化得到92.8g產品,純度達到98%,兩步總產率為60%。
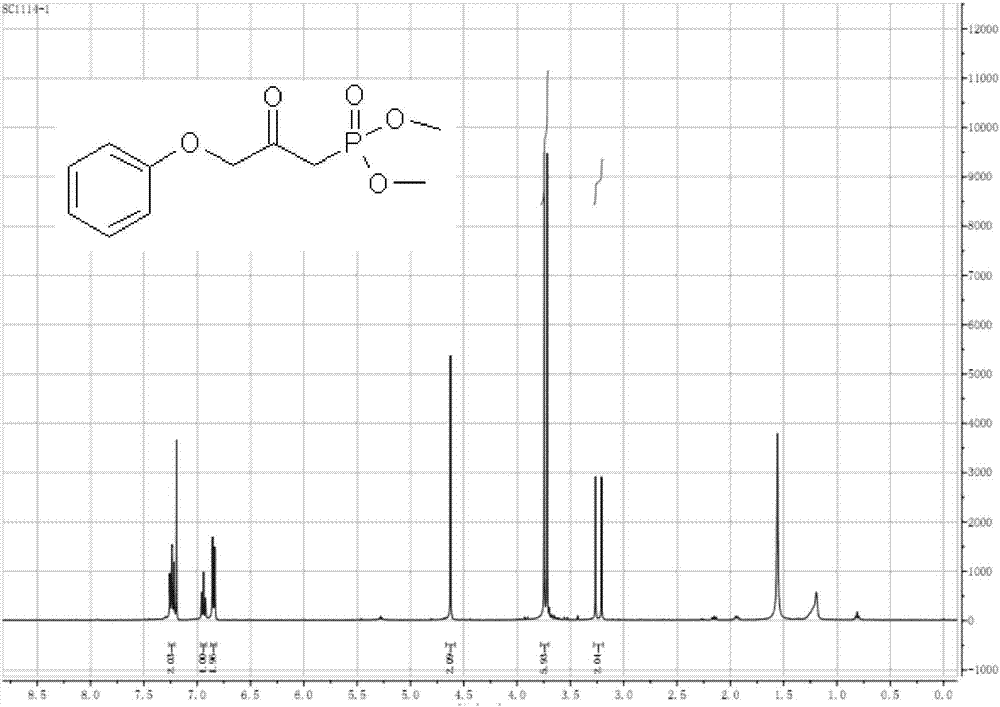
1hnmr[400mhz,cdcl3]δppm:7.32(m,2h),7.05(m,h),6.92(m,2h),4.70(s,2h),3.77(s,3h),3.75(s,3h),3.30(d,m,j=23.2hz,2h)。
實施例3
本實施例采用以下合成路線進行制備:
500毫升三口燒瓶中,20g4-苯基-1-丁烯(151mmol)溶解于250ml二氯甲烷中,冰水浴冷卻下,分批加入43.5g間氯過氧苯甲酸(151mmol),隨后室溫攪拌過夜。反應液用飽和碳酸氫鈉水溶液洗滌,干燥濃縮得到22.3g無色油狀環氧產物,產率100%,核磁比較純,直接使用于下一步。
在250ml燒瓶中,分別加入14.8g上一步產品(100mmol,1eq.),37.2g亞磷酸三甲酯(300mmol,3eq.)和120ml甲苯。反應體系加熱到65~70℃,在攪拌下,分批少量加入13.7g氯化鋅(100mmol,1eq.),然后加熱攪拌約4小時,原料反應完畢。冷卻后,減壓蒸餾將甲苯等溶劑濃縮至油狀物,加入二氯甲烷(100ml)和稀鹽酸(10%,70ml),分層,水相用二氯甲烷(100ml)反萃,合并二氯甲烷萃取液并濃縮得到油狀產物粗品,不經分離純化直接使用于下一步。
上一步粗產品(按理論值計算,100mmol,1.0eq.)溶解在200ml丙酮中,然后冰水浴下2小時內滴加40mljones試劑(8n-cro3:10.7g溶于8.8ml濃硫酸中,然后加入31.2ml水稀釋得到40ml溶液)(110mmol,1.1eq.),在反應體系攪拌5h后,檢測原料反應完全,加入7ml異丙醇,攪拌3小時后,過濾,丙酮洗滌,減壓蒸餾溶劑。加入60ml水,經二氯甲烷萃取(80ml*2),然后有機相濃縮得到粗產品,最終通過柱層析純化得到15.8g產品,純度達到98%,兩步總產率為62%。
1hnmr[400mhz,cdcl3]δppm:7.24~7.35(m,5h),3.80(s,3h),.3.78(s,3h),2.95~3.10(m,6h)。
實施例4
本實施例采用以下合成路線進行制備:
具體操作包括以下步驟:
在500ml三口瓶中,分別加入1.31g3-三氟甲基苯氧基環氧化合物(合成如實施例1,6mmol,1eq.),1.50g亞磷酸三乙酯(9mmol,1.5eq.)和0.178g氯化鋅(1.2mmol,0.2eq.),然后反應體系加熱到60℃在氬氣下攪拌過夜(大約16小時),液相檢測有少量原料(約5%~10%)剩余。冷卻反應體系至室溫,加入50ml二氯甲烷溶解,然后分別用飽和碳酸氫鈉水溶液(20ml)和飽和食鹽水(15ml)洗滌。無水硫酸鈉干燥,濃縮得粗產品,不經分離純化直接使用于下一步。由于粗品包含磷酸三乙酯和未反應完的亞磷酸三乙酯,因而產品產率大于理論值。
1hnmr[400mhz,cdcl3]δppm:7.41(t,j=8.0hz,1h),7.23(d,j=7.6hz,1h),7.14(s,1h),7.10(d,j=8.4hz,1h),4.43(m,1h),4.20(m,4h),4.16(m,2h),3.73(br,s,1h,-oh),2.24(m,2h),1.38(m,6h)。
250ml三口瓶中,2.35g上一步粗產品(按理論值計算,應為1.97g,6mmol,1.0eq.)溶解在50ml丙酮中,然后室溫下5分鐘內滴加3mljones試劑(8n-cro3:5.33g溶于4.4ml濃硫酸中,然后加入15.6ml水稀釋得到20ml溶液)(24mmol,4eq.),三乙胺(60.75mmol,1.5eq.)。
在反應體系攪拌4h后,液相檢測原料反應完畢。然后加入4ml異丙醇,攪拌1小時后,過濾,二氯甲烷洗滌,減壓蒸餾除去二氯甲烷、丙酮及異丙醇溶劑。加入30ml水,經二氯甲烷萃取(30ml*2),然后有機層用水溶液(20ml)。無水硫酸鈉干燥,濃縮得到粗產品(1.8g)。考慮到雜質極性相對很小,而產品極性卻非常大,因而純化相對非常容易。
最終經短柱層析(開始用ea/pe=1:2沖/洗,小極性雜質很快除掉,隨后用ea/meoh=9:1沖洗給出產品)純化得到1.38g產品,產率為70%。
1hnmr[400mhz,cdcl3]δppm:7.42(t,j=8.0hz,1h),7.26(d,j=6.8hz,1h),7.14(s,1h),7.11(d,j=8.4hz,1h),4.81(s,2h),4.21(m,4h),3.30(d,m,j=23.2hz,2h),1.34(m,6h)。
盡管已用具體實施例來說明和描述了本發明,然而應意識到,以上各實施例僅用以說明本發明的技術方案,而非對其限制;本領域的普通技術人員應當理解:在不背離本發明的精神和范圍的情況下,可以對前述各實施例所記載的技術方案進行修改,或者對其中部分或者全部技術特征進行等同替換;而這些修改或者替換,并不使相應技術方案的本質脫離本發明各實施例技術方案的范圍;因此,這意味著在所附權利要求中包括屬于本發明范圍內的所有這些替換和修改。
- 還沒有人留言評論。精彩留言會獲得點贊!