一種新型的交聯型堿性聚芳醚陰離子交換膜及其制備方法與應用與流程
本發明屬于離子交換膜材料領域,具體涉及一種交聯型的聚芳醚化合物、其陰離子交換膜及其制備方法與應用。
背景技術:
燃料電池是一種能夠直接將儲存在燃料以及氧化劑中的化學能轉化為電能的發電裝置,其排放產物僅為二氧化碳和水,較為清潔環保。因此,燃料電池被認為是二十一世紀最具發展前景的能量轉換設備之一。質子交換膜燃料電池(PEMFCs),作為燃料電池的一種,具有壽命長、環境友好、啟動迅速、比功率和比能量高、結構簡單的燃料電池設備。由于質子交換膜燃料電池需使用價格昂貴的全氟磺酸型聚合物(Nafion系列膜,美國杜邦)作為其質子交換膜,以貴金屬鉑-碳或鉑-釕/碳作為主要催化劑,極大地增加了生產成本,而且還存在甲醇燃料滲透以及中間產物導致電極催化劑中毒等問題,嚴重阻礙了其進一步大規模的市場化應用。
為了克服上述技術問題,研究人員提出了一種新的燃料電池技術,堿性陰離子交換膜燃料電池(AAEMFCs),它是一種在堿性環境下工作的燃料電池技術,與質子交換膜燃料電池相比,具有明顯的特點和優勢:(1)采用固體電解質膜取代傳統的液體電解質,能夠有效地阻隔陰陽兩極的燃料和氧化劑,甲醇滲透率低;(2)具有比在質子交換膜燃料電池中更快的反應速度;(3)不會造成電極催化劑中毒的情況產生;(4)可使用Ag,Ni,Co等地球儲量豐富的過渡金屬,避免了必須使用貴金屬催化劑的情況,降低了生產成本。
陰離子交換膜(AEM)作為AAEMFCs的核心組成部分,起著傳導OH-形成電流回路,并且阻隔電池正負極防止燃料滲透的作用。理想的AAEMFCs陰離子交換膜,應該具有如下特點:較低的燃料滲透率,以保證最大程度上提高電流效率;較高的OH-電導率和機械性能;優良的熱穩定性和堿性穩定性,以保證膜具有較長的使用壽命;盡可能薄的厚度(50~80μm),以降低膜的吸水率;較低的生產成本。
OH-電導率偏低與堿性穩定性較差是目前AAEMFCs中陰離子交換膜面臨的主要問題。能否制備出含有高OH-電導率的同時保持適當的溶脹度、足夠的機械強度和化學穩定性并能夠耐80℃及以上的高溫是AAEMFCs陰離子交換膜商業化面臨的重要挑戰之一。如何通過有序合理的聚合物分子主鏈與高耐堿性的側鏈設計,制備出具有高離子電導率、良好耐堿性以及較長使用壽命的陰離子交換膜是當前的一個主要研究熱點。陰離子交換膜的主鏈一般由聚芳醚類化合物或脂肪鏈聚合物構成,側基由帶電荷的季銨鹽基團構成。聚芳醚化合物具有卓越的熱穩定性、機械性能和耐腐蝕性能,在燃料電池離子交換膜材料領域受到廣泛的應用。
交聯是改善陰離子膜性能的比較好的方法,能夠顯著降低膜的吸水率和溶脹度,提高膜的熱穩定性以及耐堿性能。因此,開發一種OH-電導率高、熱穩定性和化學穩定性好、低成本的交聯型陰離子交換膜具有非常重要的實用意義。通過添加交聯劑發生化學反應使線型聚合物發生交聯形成體型聚合物是交聯的有效方法之一。
Wang等(J.Wang,et al.,J.Membrane Sci.,2012,205,5-416)報道了采用二氯苯作為交聯劑與聚醚酮反應發生化學交聯;Lee(M.S.Lee,et al.,J.Mater.Chem.,2012,22,13928-13931)以1,4-雙丙烯酰哌嗪作為交聯劑,制備的陰離子膜在堿性穩定性以及熱穩定性方面都得到了較大的提高。
本發明提供了一種交聯型的聚芳醚化合物,具有比較高的熱穩定性,其陰離子交換膜具有比較高的離子傳導率、機械性能、化學穩定性以及熱穩定性能,能夠作為陰離子交換膜應用在堿性陰離子交換膜燃料電池中。
技術實現要素:
本發明的目的之一在于提供交聯型的聚芳醚化合物的陰離子交換膜,該交聯型的陰離子交換膜具有比較高的熱穩定性、化學穩定性、良好的機械性能以及比較高的離子電導率。
本發明的目的之二在于提供上述交聯型的陰離子交換膜的制備方法。
本發明的目的之三在于提供上述交聯型的陰離子交換膜的應用。
本發明的技術方案如下:
一種新型的交聯型堿性聚芳醚陰離子交換膜,該交換膜的材料為交聯型的聚芳醚化合物,交聯型的聚芳醚化合物具有以下結構式:
其中,x和y為聚合度,x=1~200且y=0~200,0<x/(x+y)≤1,0≤y/(x+y)≤1,交聯度=a/x,聚合物的相對分子量為10000~100000;
Q為添加交聯劑T后形成的交聯單元,為如下結構式(a)~式(e)中的任意一種:
A:當y>0時:選自結構式(1)~式(3)中的任意一種,
其中R1為CH3,
中的任意一種。
選自式(4)~式(5)中的任意一種:
對應式(1)~式(3)中的結構式,分別選自結構式(6)~式(8)中的任意一種:
其中R2=CH3,中的任意一種。
選自式(9)~式(14)中的任意一種:
B:當y=0時,選自式(15)~式(16)中的任意一種:
其中R3=H,中的任意一種。
選自式(4)~式(5)中的任意一種;
對應式(15)~式(16)中的結構式,分別選自式(17)~式(18)中的任意一種:
按照本發明,所述式(Ⅰ)可分為無規共聚物和均聚物兩種情況。
通過控制投料的比例,可以控制x和y組分的比值,x和y的數值反映出聚芳醚化合物的分子量及其分子量分布范圍。
所述新型的交聯型堿性聚芳醚陰離子交換膜的厚度為50~80μm,拉伸強度為20.0~50.0MPa,膜的熱穩定性溫度為250~400℃,30℃下的OH-電導率為20.0~50.0mS·cm-1,80℃下的OH-電導率為60.0~100.0mS·cm-1,在60℃、2M NaOH溶液中放置30天后,30℃下的OH-電導率為10.0~40.0mS·cm-1。
所述的新型的交聯型堿性聚芳醚陰離子交換膜的制備方法包括以下步驟:
①當y>0時且聚合物為無規共聚物時:
步驟(1)、聚芳醚化合物的制備:將雙酚單體Ar1,雙酚芳香單體Ar3,含有鹵素原子的芳香單體Ar2和催化劑按照摩爾比1:(0.1~10):(1.1~11):(2.2~22)加入到反應容器中,然后加入帶水劑和極性非質子溶劑p,140℃下反應2~3h,后升溫至150~210℃反應3~16h,將反應物倒入乙醇中析出,并用乙醇和去離子水反復洗滌3次,80℃下真空干燥24h,得到聚芳醚化合物;所述的帶水劑與極性非質子溶劑p的體積比為(0.5~1.5):1;
所述芳香雙酚單體Ar1和Ar3具有如下結構特征:
其中選自結構式(19)~式(21)中的任意一種:
選自結構式(22)~式(27)中的任意一種:
含有鹵素原子的芳香單體Ar2具有如下結構特征:且選自結構式(28)~式(29)中的任意一種:
步驟(2)、聚芳醚化合物的溴甲基化:取步驟(1)中制備的無規聚芳醚化合物溶解在四氯乙烷中,按照聚芳醚化合物、溴化劑N-溴代丁二酰亞胺和引發劑偶氮二異丁腈摩爾比為1:(2~5):(0.1~0.25)投料,并且體系在氮氣保護下升溫至80~85℃反應5~8h,倒入乙醇中沉淀,然后用乙醇反復洗滌3次,60℃真空干燥得到溴甲基化的聚芳醚化合物;
所述的無規聚芳醚化合物具有如下結構特征:
其中,選自結構式(30)~式(32)中的任意一種,
選自式(4)~式(5)中的任意一種;
選自式(9)~式(14)中的任意一種;
步驟(3)、交聯:將上述步驟(2)中取得的溴甲基化的聚芳醚化合物溶解在極性非質子溶劑p中,形成質量分數為(8~10)wt%的溶液,加入交聯劑T,反應0.5~3h,得到新型的交聯的聚芳醚化合物。
所述的溴甲基化的聚芳醚化合物具有如下結構特征:
其中Ar1″選自式(33)~式(35)中的任意一種:
其中R4=CH2Br或CH3。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入季銨化試劑,反應20~24h,之后將溶液澆鑄在鍍膜機上洗干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入堿液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到新型的交聯型堿性聚芳醚陰離子交換膜,膜的交聯度為5%~10%。
優選的,步驟(1)中所述的催化劑為碳酸鉀、碳酸鈉或碳酸銫中的一種,所述的帶水劑為甲苯。
優選的,步驟(1)中所述的乙醇和去離子水的體積比為1:1;
優選的,步驟(1)和步驟(2)中所述的極性溶劑p可為N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、環丁砜、N-甲基吡咯烷酮、或二甲基亞砜中的任意一種。
步驟(3)中所述的交聯試劑為T,可為式(f)~式(j)中的任意一種。
步驟(4)中所述的季銨化試劑可為中的任意一種。
步驟(5)中所述的堿液為1M的氫氧化鈉(NaOH)或氫氧化鉀(KOH)溶液中的任意一種。
②當y=0且聚合物為均聚物時:
步驟(1)、聚芳醚化合物的制備:將芳香雙酚單體Ar1和含鹵素原子的芳香單體Ar2以及催化劑按照摩爾比例為1:1:(2~2.5)的比例加入到反應容器中,然后加入帶水劑和極性非質子溶劑p,140℃下反應2~3h后,升溫至150~210℃反應3~16h,將反應物倒入乙醇中析出,并用乙醇和去離子水反復洗滌3次,80℃下真空干燥24h,得到聚芳醚化合物;所述的帶水劑與極性非質子溶劑p的體積比為(0.5~1.5):1;
所述芳香雙酚單體Ar1具有如下結構特征:且選自結構式(22)或式(24)中的任意一種:
含鹵素原子的芳香單體Ar2具有如下結構特征:且選自結構式(28)~式(29)中的任意一種:
步驟(2)、聚芳醚化合物的氯甲基化:將聚芳醚化合物首先溶解在四氯乙烷中,然后將聚芳醚化合物、四氯乙烷、四氯化錫和氯甲醚按照質量比為1:(10~20):(0.1~0.2):(0.5~1)的比例加入反應容器中,50℃下反應5~10h,之后將反應溶液倒入乙醇中沉淀,得到絮狀產物,用乙醇反復洗滌后60℃下真空干燥得到氯甲基化的聚芳醚化合物;
所述的聚芳醚化合物具有如下式(Ⅳ)所示結構特征:
其中分別選自式(9)或式(11)中的任意一種:
選自式(4)~式(5)中的任意一種;
步驟(3)、交聯:將上述步驟(2)中取得的氯甲基化的聚芳醚化合物溶解在極性非質子溶劑p中,形成質量分數為(8~10)wt%的溶液,加入交聯劑T,反應0.5~3h,得到新型的交聯的聚芳醚化合物。
所述的氯甲基化的聚芳醚化合物具有如下結構特征:
其中Ar1″選自式(36)或式(37)中的任意一種:
其中R5=H或CH2Cl;
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入季銨化試劑,反應20~24h,之后將溶液澆鑄在鍍膜機上洗干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入堿液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到新型的交聯型堿性聚芳醚陰離子交換膜,膜的交聯度為5%~10%。
優選的,步驟(1)中所述的催化劑為碳酸鉀、碳酸鈉或碳酸銫中的一種,所述的帶水劑為甲苯。
優選的,步驟(1)和步驟(2)中所述的極性溶劑p可為N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、環丁砜、N-甲基吡咯烷酮、或二甲基亞砜中的任意一種。
步驟(3)中所述的交聯試劑為T,可為式(f)~式(j)中的任意一種。
步驟(4)中所述的季銨化試劑可為
中的任意一種。
步驟(5)中所述的堿液為1M的氫氧化鈉(NaOH)或氫氧化鉀(KOH)溶液中的任意一種。
本發明方法所述的制備方法和應用為優化的方案,本發明所述的反應單體、溫度、時間和其他相關反應條件均為本專利所要保護的內容,本專利所要保護的并不僅僅局限于此。
所述的新型交聯型的陰離子交換膜可以作為堿性陰離子交換膜用在堿性燃料電池中,還可以是全釩液流電池以及電滲析等領域中的應用。
與現有的技術相比,本發明具有以下優點和有益效果:
(1)本發明聚芳醚化合物主鏈分子中含有較多的芳香苯環和醚鍵,提高了聚合物的強度又增加了分子的柔軟性,采用冠醚胺或穴醚胺作為新型的交聯化試劑,由于冠醚胺或穴醚胺具有比較大的環狀結構,不僅能夠儲存較多的水分子和OH-以提高其陰離子交換膜的OH-電導率,并且能夠在一定程度上阻擋外部堿性環境中OH-的進攻,提高膜的堿性穩定性。
(2)本發明通過加入交聯劑使膜發生交聯形成具有三維網絡結構的堿性聚芳醚陰離子交換膜,提高了膜的結構穩定性,并且可以通過調節溴化度或氯甲基化程度以及交聯時間的長短來控制膜的交聯度,反應時間短且易于操作。
(3)本發明制備的新型的交聯型堿性聚芳醚陰離子交換膜具有比較高的離子電導率,30℃下OH-電導率為20.0~50.0mS·cm-1,80℃下的OH-電導率為60.0~100.0mS·cm-1,通過交聯改善了膜的吸水率和溶脹度,并且有效地提高了膜的熱穩定性和化學穩定性,可以作為陰離子交換膜應用于堿性燃料電池,同時還可以將膜交換成Cl-形式,應用到全釩液流電池中,具有比較好的應用前景。
附圖說明
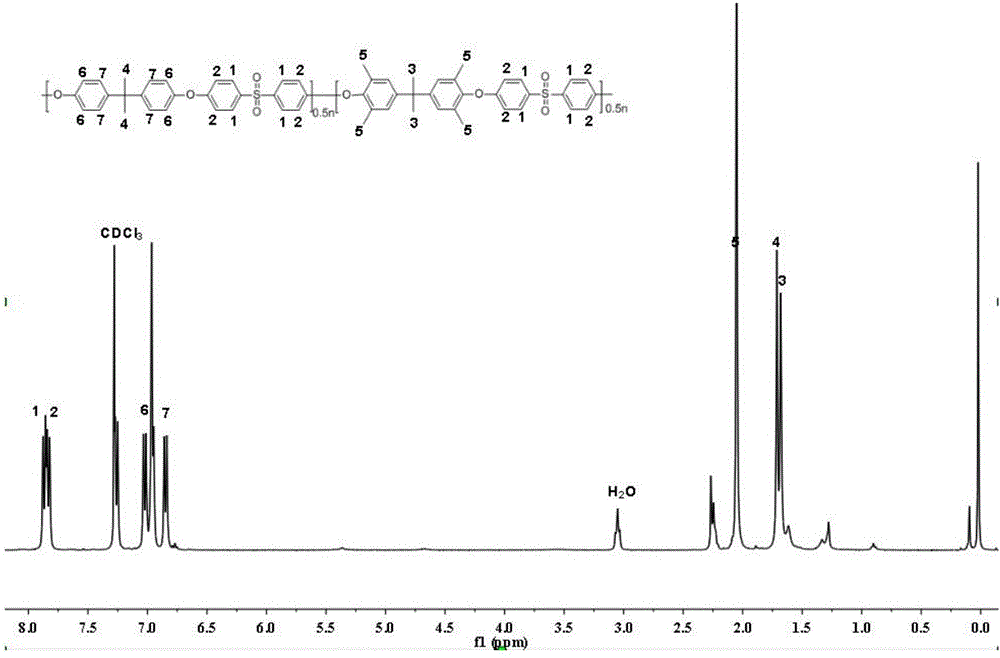
圖1為本發明的實施例1制備的無規聚芳醚化合物的核磁譜圖。
圖2為本發明的實施例1制備的溴化聚芳醚化合物核磁譜圖。
圖3為本發明的實施例1制備的新型的交聯型堿性聚芳醚陰離子交換膜的核磁譜圖。
圖4為本發明的實施例1制備的新型的交聯型堿性聚芳醚陰離子交換膜在去離子水中的OH-電導率隨溫度的變化曲線。
圖5為本發明的實施例1制備的新型的交聯型堿性聚芳醚陰離子交換膜的在2M NaOH溶液中放置30天的OH-電導率變化曲線。
具體實施方式
下面結合具體實施例對本發明作進一步的解釋說明,但本發明的實施方式并不僅僅限于此。
采用如下步驟制備新型的交聯型堿性聚芳醚陰離子交換膜:
當y>0且聚合物為無規共聚物時:新型的交聯型堿性聚芳醚化合物及其陰離子交換膜的實施案例如下:
實施例1
(1)、無規聚芳醚化合物的制備:10mmol的四甲基雙酚A(式19),1mmol的雙酚A(式22),11mmol的4,4’-二氟二苯砜(式28),加入22mmol的碳酸鉀,30mL的甲苯和20mL的環丁砜,氮氣保護,140℃下帶水2h,之后升溫至210℃反應3h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,如圖1所示,GPC測得所制備的無規聚芳醚化合物的分子量為68kg/mol,結構如下所示:
(2)、溴甲基化:取0.01mmol的無規聚芳醚,溶于20mL的1,1,2,2,-四氯乙烷中,加入0.035mmol的N-溴代丁二酰亞胺和0.0023mmol的偶氮二異丁腈,氮氣保護,80℃下反應8h,將反應液冷卻后倒入甲醇中攪拌析出,得到絮狀產物,并采用甲醇多次洗滌后60℃下真空干燥24h,得到溴甲基化的聚芳醚化合物,經過核磁共振譜檢測,如圖2所示,所制備的溴甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(33),R4=CH3或CH2Br,Ar3為式(9)
步驟(3)、交聯:取1g的上述步驟(2)中取得的溴甲基化的聚芳醚化合物溶解在9g的N-甲基吡咯烷酮中,形成質量分數為10wt%的溶液,加入交聯劑(f),反應0.5h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入1,2-二甲基咪唑作為季銨化試劑反應20h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型的交聯型堿性聚芳醚陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為80um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,在3300~3500cm-1處出現結合水與季銨鹽基團中-OH的伸縮振動峰,說明離子化和堿化成功進行,所制備的陰離子交換膜的核磁譜圖如圖3所示,結合核磁和紅外我們可以得出制備的新型交聯型的陰離子交換膜具有如下結構:
其中為式(1),為式(6),為式(9),R1=CH3或R2=CH3或Q為式(a)。
X=60,y=84,a可通過“交聯度=a/x”計算,本實施例中a=3;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜相關性能的測試:膜的吸水率(WU%)=(Wwet-Wdry)/Wdry x 100%,Wwet和Wdry分別為濕態和干態下膜的重量;溶脹度(SR%)=(KwetLwet-KdryLdry)/KdryLdry x 100%,其中Kwet和Lwet分別為膜濕態下的長度和寬度,Kdry和Ldry分別為膜干態下的長度和寬度。交聯度(%)=100%x(IEC0-IEC1)/IEC0,IEC0和IEC1分別為沒交聯和交聯后的膜的離子交換容量。
膜性能的測試結果:膜交聯前的離子交換容量IEC0=1.72meq·g-1,發生交聯后的離子交換容量IEC1=1.63meq·g-1,交聯度為5%,30℃和60℃下的吸水率分別為45.6%和81.2%,陰離子膜在去離子水中20℃~80℃的OH-電導率如圖4所示,30℃下的OH-電導率為33.4mS·cm-1,80℃下的OH-電導率為86.9mS·cm-1。膜的拉伸強度在28.3MPa,斷裂伸長率為6.7%,將該膜浸入2M NaOH溶液中60℃下放置30天的堿性穩定性曲線如圖5所示,膜在30℃下的OH-電導率仍為26.2mS·cm-1,顯示出具有良好的耐堿性穩定性。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。
實施例2
(1)無規聚芳醚化合物的制備:3mmol四甲基聯苯二酚(式20),7mmol雙酚AF(式23),10mmol 4,4’-二氟二苯砜(式28),加入20mmol的碳酸鈉,20mL的甲苯和20mL的N,N-二甲基乙酰胺,氮氣保護,140℃下帶水2.5h,之后升溫至165℃反應12h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,所制備的無規聚芳醚化合物的分子量為62kg/mol,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,結構如下:
式中,Ar1為式(20),Ar3為式(10)
(2)溴甲基化:取0.01mmol(0.62g)的無規聚芳醚,溶于20mL的1,1,2,2,-四氯乙烷中,加入0.03mmol的N-溴代丁二酰亞胺和0.002mmol的偶氮二異丁腈,氮氣保護,85℃下反應5h,將反應液冷卻后倒入甲醇中攪拌析出,得到絮狀產物,并采用甲醇多次洗滌后60℃下真空干燥24h,得到溴甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的溴甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(34),R1=CH3或CH2Br,Ar3為式(10)
步驟(3)、交聯:取1g的上述步驟(2)中取得的溴甲基化的聚芳醚化合物溶解在11.5g的N,N-二甲基乙酰胺中,形成質量分數為8wt%的溶液,加入交聯化試劑(式g),反應2h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入N-甲基咪唑作為季銨化試劑反應22h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為78um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(2),為式(7),R1=CH3或為式(10),R2=CH3或Q為式(b)。
膜相關性能的測試:同上;
X=29,y=88,a可通過“交聯度=a/x”計算,本實施例中a=2.5;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜交聯前的離子交換容量IEC0=1.69meq·g-1,發生交聯后的離子交換容量IEC1=1.56meq·g-1,交聯度為8.6%,30℃和60℃下的吸水率分別為35.6%和76.8%,30℃下的OH-電導率為32.7mS·cm-1,80℃下的OH-電導率為73.6mS·cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在39.2MPa,斷裂伸長率為8.7%,將該膜浸入2M NaOH溶液中60℃下放置7天后,膜在30℃下的OH-電導率仍為28.3mS·cm-1。
實施例3
(1)無規聚芳醚化合物的制備:2mmol四甲基雙酚芴(式21),8mmol對羥基連苯二酚(式24),4,4’-二氟二苯砜(10mmol),加入20mmol的碳酸銫,10mL的甲苯和20mL的N,N-二甲基甲酰胺,氮氣保護,140℃下帶水3h,之后升溫至150℃反應16h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,所制備的無規聚芳醚化合物的分子量為52kg/mol,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,結構如下:
式中,Ar1為式(32),Ar3為式(11)
(3)溴甲基化:取0.01mmol(0.52g)的無規聚芳醚,溶于20mL的1,1,2,2,-四氯乙烷中,加入0.04mmol的N-溴代丁二酰亞胺和0.025mmol的偶氮二異丁腈,氮氣保護,84℃下反應7h,將反應液冷卻后倒入甲醇中攪拌析出,得到絮狀產物,并采用甲醇多次洗滌后60℃下真空干燥24h,得到溴甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的溴甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(21),R4=CH3或CH2Br,Ar3為式(11)
步驟(3)、交聯:取1g的上述步驟(2)中取得的溴甲基化的聚芳醚化合物溶解在10.11g的N,N-二甲基甲酰胺中,形成質量分數為9wt%的溶液,加入交聯化試劑(式h),反應2.5h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入吡啶作為季銨化試劑反應23h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的NaOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為65um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(3),為式(6),R1=CH3或為式(11),R2=CH3或Q為式(c)。
膜相關性能的測試:同上;
X=30,y=84,a可通過“交聯度=a/x”計算,本實施例中a=2.7;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.83meq g-1,膜發生交聯后的離子交換容量IEC1=1.67meq g-1,交聯度為9%,30℃和60℃下的吸水率分別為41.2%和86.3%,30℃下的OH-電導率為45.6mS cm-1,80℃下的OH-電導率為93.2mS cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在25.8MPa,斷裂伸長率12.4%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為37.5mS cm-1。
實施例4
(1)無規聚芳醚化合物的制備:4mmol四甲基雙酚A(式19),6mmol的4,4’-二羥基二苯砜(式25),10mmol 4,4’-二氟二苯酮(式29),加入20mmol的碳酸鉀,20mL的甲苯和20mL的二甲基亞砜,氮氣保護,140℃下帶水2.2h,之后升溫至180℃反應8h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,所制備的無規聚芳醚化合物的分子量為63kg/mol,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,結構如下:
式中,Ar1為式(30),Ar3為式(12)
(4)溴甲基化:取0.01mmol的無規聚芳醚,溶于20mL的1,1,2,2,-四氯乙烷中,加入0.05mmol的N-溴代丁二酰亞胺和0.0025mmol的偶氮二異丁腈,氮氣保護,85℃下反應8h,將反應液冷卻后倒入甲醇中攪拌析出,得到絮狀產物,并采用甲醇多次洗滌后60℃下真空干燥24h,得到溴甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的溴甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(33),R4=CH3或CH2Br,Ar3為式(12)
步驟(3)、交聯:取1g的上述步驟(2)中取得的溴甲基化的聚芳醚化合物溶解在11.5g的環丁砜中,形成質量分數為8wt%的溶液,加入交聯化試劑(式i),反應3h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入胍作為季銨化試劑反應24h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為60um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(1),為式(4),R1=CH3或為式(13),R2=CH3或Q為式(d)。
膜相關性能的測試:同上;
X=56,y=73,a可通過“交聯度=a/x”計算,本實施例中a=5.6;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC=1.92meq·g-1,膜發生交聯后的離子交換容量IEC=1.73meq·g-1,交聯度為10%,30℃和60℃下的吸水率分別為56.8%和104.3%,30℃下的OH-電導率為50.0mS·cm-1,80℃下的OH-電導率為100.0mS·cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在20.0MPa,斷裂伸長率為16.3%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為40.0mS·cm-1。
實施例5
(1)無規聚芳醚化合物的制備:5mmol四甲基聯苯二酚(式20),5mmol 4,4’-二羥基二苯酮(式26),10mmol 4,4’-二氟二苯酮(式29),加入25mmol的碳酸鉀,25mL的甲苯和20mL的N-甲基吡咯烷酮,氮氣保護,150℃下帶水3h,之后升溫至200℃反應6h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,所制備的無規聚芳醚化合物的分子量為56kg/mol,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,結構如下:
式中,Ar1為式(20),Ar3為式(13)
(2)溴甲基化:取0.01mmol(0.68g)的無規聚芳醚,溶于20mL的1,1,2,2,-四氯乙烷中,加入0.02mmol的N-溴代丁二酰亞胺和0.001mmol的偶氮二異丁腈,氮氣保護,80℃下反應5h,將反應液冷卻后倒入甲醇中攪拌析出,得到絮狀產物,并采用甲醇多次洗滌后60℃下真空干燥24h,得到溴甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的溴甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(34),R4=CH3或CH2Br,Ar3為式(13)
步驟(3)、交聯:取1g的上述步驟(2)中取得的溴甲基化的聚芳醚化合物溶解在9g的N,N-二甲基乙酰胺中,形成質量分數為10wt%的溶液,加入交聯化試劑(式j),反應1.5h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入三甲胺作為季銨化試劑反應24h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的NaOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為50um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(2),為式(5),R1=CH3或為式(13),R2=CH3或Q為式(e)。
膜相關性能的測試:同上;
X=66,y=66,a可通過“交聯度=a/x”計算,本實施例中a=4.5;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.46meq g-1,膜發生交聯前的離子交換容量IEC10=1.36meq g-1,交聯度為6.8%,30℃和60℃下的吸水率分別為32.2%和58.5%,30℃下的OH-電導率為20.0mS cm-1,80℃下的OH-電導率為60.0mS cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在47.7MPa,斷裂伸長率為6.9%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為10.0mS cm-1。
實施例6
(1)無規聚芳醚化合物的制備:1mmol四甲基雙酚芴(式21),10mmol的雙酚環己烷(式27),11mmol的4,4’-二氟二苯酮(式29),加入22mmol的碳酸銫,15mL的甲苯和20mL的環丁砜,氮氣保護,140℃下帶水3h,之后升溫至210℃反應3h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,所制備的無規聚芳醚化合物的分子量為61kg/mol,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,結構如下:
式中,Ar1為式(32),Ar3為式(14)
(2)溴甲基化:取0.01mmol的無規聚芳醚,溶于20mL的1,1,2,2,-四氯乙烷中,加入0.025mmol的N-溴代丁二酰亞胺和0.0015mmol的偶氮二異丁腈,氮氣保護,82℃下反應6h,將反應液冷卻后倒入甲醇中攪拌析出,得到絮狀產物,并采用甲醇多次洗滌后60℃下真空干燥24h,得到溴甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的溴甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(35),R4=CH3或CH2Br,Ar3為式(14)
步驟(3)、交聯:取1g的上述步驟(2)中取得的溴甲基化的聚芳醚化合物溶解在11.5g的二甲基亞砜中,形成質量分數為8wt%的溶液,加入交聯化試劑(式f),反應0.8h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入三苯基膦作為季銨化試劑反應24h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為69um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(3),為式(6),R1=CH3或為式(13),R2=CH3或Q為式(a)。
膜相關性能的測試:同上;
X=82,y=31,a可通過“交聯度=a/x”計算,本實施例中a=4.51;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.62meq g-1,膜發生交聯后的離子交換容量IEC1=1.53meq g-1,交聯度為5.5%,30℃和60℃下的吸水率分別為45.6%和87.8%,30℃下的OH-電導率為28.8mS cm-1,80℃下的OH-電導率為65.6mS cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在42.5MPa,斷裂伸長率為6.8%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為20.1mS cm-1。
當y=0且聚合物為均聚物時,新型交聯的聚芳醚化合物及其陰離子交換膜的實施案例如下:
實施例7
(1)無規聚芳醚化合物的制備:10mmol的雙酚A(式22),10mmol的4,4’-二氟二苯砜(式28),加入20mmol的碳酸鉀,30mL的甲苯和20mL的環丁砜,氮氣保護,140℃下帶水2h,之后升溫至210℃反應3h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,所制備的無規聚芳醚化合物的分子量為57kg/mol,結構如下所示:
式中,Ar1為式(9),Ar2為式(4)
(2)氯甲基化:取10g的聚芳醚化合物,溶于100g的1,1,2,2,-四氯乙烷中,加入1.2g的四氯化錫(SnCl4)和6g的氯甲醚,氮氣保護,50℃下反應6h,將反應液冷卻后倒入乙醇中攪拌析出,得到絮狀產物,并采用乙醇多次洗滌后60℃下真空干燥24h,得到氯甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的氯甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(36),R5=H或CH2Cl,Ar2為式(4)
步驟(3)、交聯:取1g的上述步驟(2)中取得的氯甲基化的聚芳醚化合物溶解在9g的N-甲基吡咯烷酮中,形成質量分數為10wt%的溶液,加入少量的交聯試劑(f),反應0.5h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入N-甲基咪唑作為季銨化試劑反應24h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到新型的交聯型堿性聚芳醚陰離子交換膜,膜的厚度為56um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,在3300~3500cm-1處出現結合水與季銨鹽基團中-OH的伸縮振動峰,說明離子化和堿化成功進行,結合核磁和紅外我們可以得出制備的新型交聯型的陰離子交換膜具有如下結構:
其中為式(15),為式(17),R1=H或R2=H或Q為式(a)。
膜相關性能的測試:膜的吸水率(WU%)=(Wwet-Wdry)/Wdry x 100%,Wwet和Wdry分別為濕態和干態下膜的重量;溶脹度(SR%)=(KwetLwet-KdryLdry)/KdryLdry x 100%,其中Kwet和Lwet分別為膜濕態下的長度和寬度,Kdry和Ldry分別為膜干態下的長度和寬度。交聯度(%)=100%x(IEC0-IEC1)/IEC0,IEC0和IEC1分別為沒交聯和交聯后的膜的離子交換容量。
X=128,a可通過“交聯度=a/x”計算,本實施例中a=6.4;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.61meq·g-1,膜發生交聯后的離子交換容量IEC1=1.52meq·g-1,交聯度為5%,30℃和60℃下的吸水率分別為35.6%和62.2%,30℃下的OH-電導率為25.8mS·cm-1,80℃下的OH-電導率為69.6mS·cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在44.1MPa,斷裂伸長率為7.3%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為17.4mS·cm-1,顯示出具有良好的耐堿性穩定性。
實施例8
(1)無規聚芳醚化合物的制備:10mmol對羥基聯苯(式24),10mmol 4,4’-二氟二苯砜(式29),加入22mmol的碳酸鈉,20mL的甲苯和20mL的N,N-二甲基乙酰胺,氮氣保護,140℃下帶水2.3h,之后升溫至165℃反應12h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,所制備的無規聚芳醚化合物的分子量為54kg/mol,結構如下所示:
式中,Ar1為式(11),Ar2為式(4)
(2)氯甲基化:取10g的均聚聚芳醚化合物,溶于140g的1,1,2,2,-四氯乙烷中,加入1.6g的四氯化錫(SnCl4)和8g的氯甲醚,氮氣保護,50℃下反應8h,將反應液冷卻后倒入乙醇中攪拌析出,得到絮狀產物,并采用乙醇多次洗滌后60℃下真空干燥24h,得到氯甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的氯甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(37),R5=H或CH2Cl,Ar2為式(4)
步驟(3)、交聯:取1g的上述步驟(2)中取得的氯甲基化的聚芳醚化合物溶解在11.5g的二甲基亞砜中,形成質量分數為8wt%的溶液,加入少量的交聯試劑,反應1.5h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入N-甲基咪唑作為季銨化試劑反應22h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為67um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,在3300~3500cm-1處出現結合水與季銨鹽基團中-OH的伸縮振動峰,說明離子化和堿化成功進行,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(16),為式(18),R1=H或R2=H或Q為式(b)。
膜相關性能的測試:膜的吸水率(WU%)=(Wwet-Wdry)/Wdry x 100%,Wwet和Wdry分別為濕態和干態下膜的重量;溶脹度(SR%)=(KwetLwet-KdryLdry)/KdryLdry x 100%,其中Kwet和Lwet分別為膜濕態下的長度和寬度,Kdry和Ldry分別為膜干態下的長度和寬度。交聯度(%)=100%x(IEC0-IEC1)/IEC0,IEC0和IEC1分別為沒交聯和交聯后的膜的離子交換容量。
X=130,a可通過“交聯度=a/x”計算,本實施例中a=8.06;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.68meq g-1,膜發生交聯前的離子交換容量IEC1=1.57meq·g-1,交聯度為6.2%,30℃和60℃下的吸水率分別為42.6%和85.2%,30℃下的OH-電導率為39.5mS·cm-1,80℃下的OH-電導率為78.6mS·cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在37.6MPa,斷裂伸長率為12.5%,將該膜浸入2M NaOH溶液中60℃下放置7天后,膜在30℃下的OH-電導率仍為33.5mS·cm-1,顯示出具有良好的耐堿性穩定性。
實施例9
(1)無規聚芳醚化合物的制備:10mmol的雙酚A(式22),10mmol的4,4’-二氟二苯酮(式29),加入25mmol的碳酸銫,10mL的甲苯和20mL的N,N-二甲基甲酰胺,氮氣保護,140℃下帶水2.5h,之后升溫至150℃反應16h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,所制備的無規聚芳醚化合物的分子量為51kg/mol,結構如下所示:
式中,Ar1為式(9),Ar2為式(5)
(2)氯甲基化:取10g的均聚聚芳醚化合物,溶于150g的1,1,2,2,-四氯乙烷中,加入1.5g的四氯化錫(SnCl4)和7g的氯甲醚,氮氣保護,50℃下反應7h,將反應液冷卻后倒入乙醇中攪拌析出,得到絮狀產物,并采用乙醇多次洗滌后60℃下真空干燥24h,得到氯甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的氯甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(36),R5=H或CH2Cl,Ar2為式(5)
步驟(3)、交聯:取1g的上述步驟(2)中取得的氯甲基化的聚芳醚化合物溶解在10.1g的環丁砜中,形成質量分數為9wt%的溶液,加入少量的季銨化試劑,反應2h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入吡啶作為季銨化試劑反應20h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為80um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,在3300~3500cm-1處出現結合水與季銨鹽基團中-OH的伸縮振動峰,說明離子化和堿化成功進行,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(15),為式(17),R1=H或R2=H或Q為式(c)。
膜相關性能的測試:膜的吸水率(WU%)=(Wwet-Wdry)/Wdry x 100%,Wwet和Wdry分別為濕態和干態下膜的重量;溶脹度(SR%)=(KwetLwet-KdryLdry)/KdryLdry x 100%,其中Kwet和Lwet分別為膜濕態下的長度和寬度,Kdry和Ldry分別為膜干態下的長度和寬度。交聯度(%)=100%x(IEC0-IEC1)/IEC0,IEC0和IEC1分別為沒交聯和交聯后的膜的離子交換容量。
X=125,a可通過“交聯度=a/x”計算,本實施例中a=9.4;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.66meq g-1,膜發生交聯后的離子交換容量IEC1=1.54meq g-1,交聯度為7.5%,30℃和60℃下的吸水率分別為47.6%和88.6%,30℃下的OH-電導率為34.6mS cm-1,80℃下的OH-電導率為72.5mS cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在41.9MPa,斷裂伸長率為10.7%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為30.6mS cm-1,顯示出具有良好的耐堿性穩定性。
實施例10
(1)無規聚芳醚化合物的制備:10mmol對羥基聯苯(式24),10mmol 4,4’-二氟二苯酮(式29),加入25mmol的碳酸鉀,10mL的甲苯和20mL的二甲基亞砜,氮氣保護,140℃下帶水3h,之后升溫至180℃反應10h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,所制備的無規聚芳醚化合物的分子量為54kg/mol,結構如下所示:
式中,Ar1為式(11),Ar2為式(5)
(2)氯甲基化:取10g的均聚聚芳醚化合物,溶于120g的1,1,2,2,-四氯乙烷中,加入1g的四氯化錫(SnCl4)和5g的氯甲醚,氮氣保護,50℃下反應5h,將反應液冷卻后倒入乙醇中攪拌析出,得到絮狀產物,并采用乙醇多次洗滌后60℃下真空干燥24h,得到氯甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的氯甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(37),R5=H或CH2Cl,Ar2為式(5)
步驟(3)、交聯:取1g的上述步驟(2)中取得的氯甲基化的聚芳醚化合物溶解在11.5g的N,N-二甲基甲酰胺中,形成質量分數為8wt%的溶液,加入交聯試劑,反應2.3h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入胍作為季銨化試劑反應24h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為72um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,在3300~3500cm-1處出現結合水與季銨鹽基團中-OH的伸縮振動峰,說明離子化和堿化成功進行,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(16),為式(18),R1=H或R2=H或Q為式(d)。
膜相關性能的測試:膜的吸水率(WU%)=(Wwet-Wdry)/Wdry x 100%,Wwet和Wdry分別為濕態和干態下膜的重量;溶脹度(SR%)=(KwetLwet-KdryLdry)/KdryLdry x 100%,其中Kwet和Lwet分別為膜濕態下的長度和寬度,Kdry和Ldry分別為膜干態下的長度和寬度。交聯度(%)=100%x(IEC0-IEC1)/IEC0,IEC0和IEC1分別為沒交聯和交聯后的膜的離子交換容量。
X=142,a可通過“交聯度=a/x”計算,本實施例中a=12.35;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.45meq·g-1,膜發生交聯后的離子交換容量IEC1=1.32meq·g-1,交聯度為8.7%,30℃和60℃下的吸水率分別為38.2%和57.9%,30℃下的OH-電導率為20.0mS·cm-1,80℃下的OH-電導率為60.0mS·cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在50.0MPa,斷裂伸長率為6.5%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為10.0mS·cm-1,顯示出具有良好的耐堿性穩定性。
實施例11
(1)無規聚芳醚化合物的制備:10mmol雙酚A(式22),10mmol 4,4’-二氟二苯砜(式28),加入25mmol的碳酸鈉,30mL的甲苯和20mL的N-甲基吡咯烷酮,氮氣保護,140℃下帶水2.8h,之后升溫至200℃反應6h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,所制備的無規聚芳醚化合物的分子量為51kg/mol,結構如下所示:
式中,Ar1為式(9),Ar2為式(4)
(2)氯甲基化:取10g的均聚聚芳醚化合物,溶于200g的1,1,2,2,-四氯乙烷中,加入1.8g的四氯化錫(SnCl4)和8g的氯甲醚,氮氣保護,50℃下反應9h,將反應液冷卻后倒入乙醇中攪拌析出,得到絮狀產物,并采用乙醇多次洗滌后60℃下真空干燥24h,得到氯甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的氯甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(36),R1=H或CH2Cl,Ar2為式(4)
步驟(3)、交聯:取1g的上述步驟(2)中取得的氯甲基化的聚芳醚化合物溶解在10.1g的N-甲基吡咯烷酮中,形成質量分數為9wt%的溶液,加入交聯試劑,反應2.6h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入三甲胺作為季銨化試劑反應23h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為50um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,在3300~3500cm-1處出現結合水與季銨鹽基團中-OH的伸縮振動峰,說明離子化和堿化成功進行,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
膜相關性能的測試:膜的吸水率(WU%)=(Wwet-Wdry)/Wdry x 100%,Wwet和Wdry分別為濕態和干態下膜的重量;溶脹度(SR%)=(KwetLwet-KdryLdry)/KdryLdry x 100%,其中Kwet和Lwet分別為膜濕態下的長度和寬度,Kdry和Ldry分別為膜干態下的長度和寬度。交聯度(%)=100%x(IEC0-IEC1)/IEC0,IEC0和IEC1分別為沒交聯和交聯后的膜的離子交換容量。
X=115,a可通過“交聯度=a/x”計算,本實施例中a=11.04;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.78meq·g-1,膜發生交聯后的離子交換容量IEC1=1.61meq·g-1,交聯度為9.6%,30℃和60℃下的吸水率分別為48.2%和96.5%,30℃下的OH-電導率為41.7mS·cm-1,80℃下的OH-電導率為89.6mS·cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在33.4MPa,斷裂伸長率為8.8%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為35.6mS·cm-1,顯示出具有良好的耐堿性穩定性。
實施例12
(1)無規聚芳醚化合物的制備:10mmol雙酚A(式24),10mmol 4,4’-二氟二苯砜(式29),加入25mmol的碳酸銫,25mL的甲苯和20mL的環丁砜,氮氣保護,140℃下帶水3h,之后升溫至210℃反應3h,將反應溶液倒入裝有300mL甲醇的燒杯中攪拌析出,并采用甲醇和去離子水和反復洗滌,80℃下真空干燥,得到灰色聚合物樹脂,并采用核磁共振氫譜對制備的聚芳醚化合物的結構進行了表征,所制備的無規聚芳醚化合物的分子量為60kg/mol,結構如下所示:
式中,Ar1為式(11),Ar2為式(5)
(2)氯甲基化:取10g的均聚聚芳醚化合物,溶于200g的1,1,2,2,-四氯乙烷中,加入2g的四氯化錫(SnCl4)和10g的氯甲醚,氮氣保護,50℃下反應10h,將反應液冷卻后倒入乙醇中攪拌析出,得到絮狀產物,并采用乙醇多次洗滌后60℃下真空干燥24h,得到氯甲基化的聚芳醚化合物,經過核磁共振譜檢測,所制備的氯甲基化的聚芳醚化合物的結構如下:
式中,Ar1″為式(37),R1=H或CH2Cl,Ar2為式(5)
步驟(3)、交聯:取1g的上述步驟(2)中取得的氯甲基化的聚芳醚化合物溶解在9g的N,N-二甲基乙酰胺中,形成質量分數為10wt%的溶液,加入交聯試劑,反應3h,得到新型的交聯的聚芳醚化合物。
步驟(4)、季銨化:往交聯的聚芳醚化合物中加入三苯基膦作為季銨化試劑反應24h,之后將溶液澆鑄在鍍膜機上干凈的玻璃板上面,50℃下干燥24h以使溶劑揮發完全,將膜從玻璃板上面揭下得到季銨化的新型交聯的陰離子交換膜。
步驟(5)、堿化:將步驟(4)中得到的新型交聯的陰離子交換膜浸入1M的KOH溶液中浸泡24h使其充分地進行離子交換,之后用去離子水將膜洗滌至中性,得到堿性的新型交聯的聚芳醚陰離子交換膜,膜的厚度為66um,首先采用核磁共振對膜的結構進行了表征,之后并采用傅里葉紅外光譜進行測試分析,在3300~3500cm-1處出現結合水與季銨鹽基團中-OH的伸縮振動峰,說明離子化和堿化成功進行,結合核磁和紅外我們可以得出制備的新型的交聯型堿性聚芳醚陰離子交換膜具有如下結構:
其中為式(16),為式(18),R3=H或Q為式(a)。
膜相關性能的測試:膜的吸水率(WU%)=(Wwet-Wdry)/Wdry x 100%,Wwet和Wdry分別為濕態和干態下膜的重量;溶脹度(SR%)=(KwetLwet-KdryLdry)/KdryLdry x 100%,其中Kwet和Lwet分別為膜濕態下的長度和寬度,Kdry和Ldry分別為膜干態下的長度和寬度。交聯度(%)=100%x(IEC0-IEC1)/IEC0,IEC0和IEC1分別為沒交聯和交聯后的膜的離子交換容量。
X=160,a可通過“交聯度=a/x”計算,本實施例中a=16;其中交聯度(%)=(IEC0-IEC1)/IEC0X 100%計算,其中IEC0和IEC1分別為發生交聯前和發生交聯后的膜的離子交換容量。
膜性能的測試結果:膜發生交聯前的離子交換容量IEC0=1.88meq·g-1,膜發生交聯后的離子交換容量IEC1=1.69meq·g-1,交聯度為10%,30℃和60℃下的吸水率分別為57.9%和123.6%,30℃下的OH-電導率為50.0cm-1,80℃下的OH-電導率為100.0mS·cm-1。采用熱失重分析TGA顯示膜的側鏈季銨鹽離子基團的降解溫度為260~280℃,而堿性燃料電池的工作溫度為室溫~80℃,顯示其具有足夠高的熱穩定性能可以應用在堿性燃料電池中。膜的拉伸強度在22.6MPa,斷裂伸長率為13.5%,將該膜浸入2M NaOH溶液中60℃下放置30天后,膜在30℃下的OH-電導率仍為40.0mS·cm-1,顯示出具有良好的耐堿性穩定性。
- 還沒有人留言評論。精彩留言會獲得點贊!