二氫雙苯并惡庚衍生物及其組合物與應用的制作方法
本發明涉及二氫雙苯并惡庚衍生物及其組合物與應用,屬于抗腫瘤藥學技術領域。
背景技術:
惡性腫瘤嚴重威脅人類的生命健康,每年全世界約有700萬人死于癌癥,約占總死亡人數的四分之一。目前我國現有癌癥患者死亡率逾30%,已成為我國居民死亡的第二大因素。藥物治療已成為對惡性腫瘤有效而又普遍使用的一種治療方法。盡管自1942年耶魯大學的Gilman等首次證明鹽酸氮芥對小鼠Gardner淋巴瘤有治療作用以來,腫瘤的藥物治療取得了長足的進展,并成為當前臨床治療不可或缺的主要措施。但高毒副作用、耐藥等問題仍然是臨床腫瘤藥物治療遇到的主要障礙。臨床上應用的抗腫瘤藥物種類繁多,其中化療藥物主要有烷化劑鉬絡合物抗腫瘤藥物、蒽環類抗腫瘤藥物、破壞DNA的抗生素等。此外,天然抗腫瘤藥物的研究也占有相當大的比例,如目前臨床上常用一些藥物有喜樹堿、長春新堿、紫杉醇等。
多聚(ADP-核糖)聚合酶(poly(ADP-ribose)polymerase-1,PARP1)是一個高度保守的酶,主要集中在DNA損傷的維護和維修,參與多種生物學過程,包括細胞凋亡,染色體的穩定性、基因擴增、轉錄調控和細胞分裂。當DNA損傷時,PARP1結合到單鏈斷裂(SSBs)位點,成為催化活性形式。它利用還原型煙酰胺腺嘌呤二核苷酸(NAD)為底物,合成直鏈和支鏈的聚(ADP-核糖)(PAR)到PARP1本身以及核靶蛋白如組蛋白拓撲異構酶,DNA聚合酶和DNA連接酶。合成的PAR鏈介導的堿基切除修復(BER)通過招募BER蛋白如XRCC1,DNA連接酶和DNA聚合酶III,β(POLβ)受損的DNA位點。抑制PARP1會導致SSBs的積累和阻礙DNA修復機制,最終導致雙鏈斷裂(DSBs)。有趣的是,在許多類型的癌癥中PARP1發生過表達,如黑色素瘤顯示,膠質母細胞瘤和乳腺癌。此外,parp1高表達與三陰性乳腺癌(TNBC)密切相關。因此,針對PARP1抑制其相關的生物學功能可以為我們提供了潛在的癌癥治療的新策略,尤其是在TNBC。
以前的研究已經報道,抑制PARP1導致合成致死性在BRCA1/2突變的卵巢癌和乳腺癌中,這使BRCA1/2基因缺失腫瘤細胞被parp1抑制劑選擇性地靶向。目前,各種各樣的PARP抑制劑,如olaparib,rucaparib,bmn-673以及Niraparib,處于不同的臨床實驗階段。從化學角度來看,PARP抑制劑的大多數化學支架含有酰胺結構,更多的新的化學結構可以在將來被發現;從生物學的角度來看,雖然這些PARP抑制劑有高PARP1/2抑制和抗腫瘤活性;但是,長期用藥會伴隨藥物的耐藥性,導致腫瘤復發和轉移。因此,除了深入的探討抑制劑耐藥機制與PARP依賴途徑和腫瘤特異性的缺陷,開發一種新型的具有結構多樣性表現出更好的療效和更好的安全性的PARP抑制劑是治療TNBC很有前途的策略。
技術實現要素:
本發明解決的技術問題是提供一種作為PARP抑制劑的新化合物。
本發明提供可如式Ⅰ所示的化合物或其藥學上可接受的鹽:
其中,R1為-H或C1~C4烷基;R2為-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。
進一步的,優選R1為-H;R2為-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。
作為優選方案,R1為-H;R2為-H、-CH3、-NH2。
進一步的,優選R1為-H;R2為-NH2。
本發明還提供上述化合物或其藥學上可接受的鹽在制備抗腫瘤藥物中的用途。
進一步的,所述抗腫瘤藥物優選為PARP抑制劑類藥物。
所述抗腫瘤藥物優選為治療三陰性乳腺癌的藥物。
本發明還提供一種藥物組合物,它是包含有效劑量的上述化合物或其藥學上可接受的鹽的制劑。
本發明制備的化合物或其藥學上可接受的鹽,可以作為PARP抑制劑,具有一定的抗腫瘤活性,能有效的抑制某些癌細胞的生長。本發明化合物對多種腫瘤細胞,特別是乳腺癌細胞具有明顯的抑制作用。
附圖說明
圖1A為用不同濃度的化合物2處理MCF-7,MDA-MB-231,MDA-MB-436,MDA-MB-468細胞系,通過MTT測定的細胞的存活率。
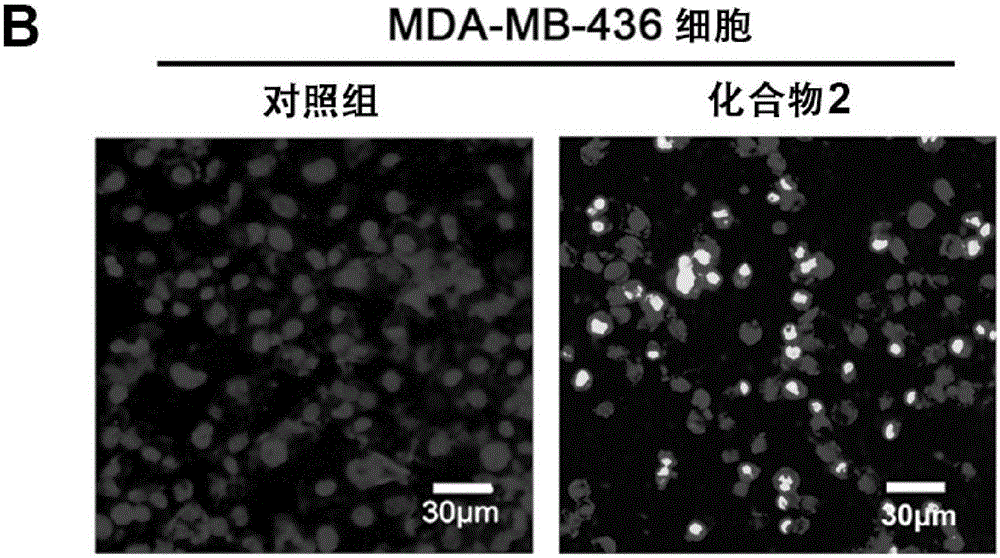
圖1B為Hoechst 33258熒光染色檢測細胞DNA損傷(Scale bar=200μm)。
圖1C為用不同濃度的化合物2處理MDA-MB-436細胞系24小時,通過流式分析測定的凋亡率。
圖1D為化合物2處理MDA-MB-436細胞,利用蛋白質印記法測定了Bax,Bcl-2,Caspase-3,PARP1和PAR的表達量。
圖1E為用化合物2處理MDA-MB-436細胞,測定其抑制細胞轉移的能力(Scale bar=100μm)。
圖2A為不同組小鼠腫瘤相對體積,**,P<0.01,與對照組相比。
圖2B為不同組小鼠相對瘤重,*,P<0.05;***,P<0.001,與對照組相比。
圖2C為.治療期間不同組的小鼠體重,**,P<0.01,與對照組相比。
圖2D為不同組小鼠的肝、脾、腎,*,P<0.05;**,P<0.01,與對照組相比。
圖2E為Ki-67和PAR的免疫組化分析(Scale bar=200μm,化合物2為高劑量組)。
圖2F為腫瘤離體組織的PARP,PAR和Caspase-3的蛋白印記分析。
具體實施方式
本發明提供可如式Ⅰ所示的化合物或其藥學上可接受的鹽:
其中,R1為-H或C1~C4烷基;R2為-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。
其中,優選R1為-H;R2為-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基。作為優選方案,R1為-H;R2為-H、-CH3、-NH2。進一步的,優選R1為-H;R2為-NH2。
其中,優選R1為-CH3,R2為-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基;更優選R1為-CH3,R2為-CH3或-CH2CH3。
其中,優選R1為-CH2CH3,R2為-H、-NH2、-CH2CH2OH、-CH2CH2CH2OCH3、或C1~C4烷基;更優選R1為--CH2CH3,R2為-CH2CH3。
下面是本發明的化合物的一些優選結構。
本發明還提供上述化合物或其藥學上可接受的鹽在制備抗腫瘤藥物中的用途。
進一步的,所述抗腫瘤藥物優選為PARP抑制劑類藥物。
所述抗腫瘤藥物優選為治療三陰性乳腺癌的藥物。
本發明還提供一種藥物組合物,它是包含有效劑量的上述化合物或其藥學上可接受的鹽的制劑。
可以通過本領域已知的方法可將本發明化合物制成以下形式:片劑、膠囊劑、水性或油性溶液劑、混懸劑、乳劑、乳膏劑、軟膏劑、凝膠劑、噴鼻劑、栓劑、用于吸入的細小分散的粉劑或氣霧劑或噴霧劑、用于胃腸道外(包括靜脈內、肌內或輸注)的無菌水性或油性溶液或混懸劑或無菌乳劑。可采用無菌水或水-丙二醇溶液作為溶劑來制備液體制劑,還可將活性組分配制在聚乙二醇水溶液中。用于口服給予的水性溶液可通過將活性組分溶解在水中并按需要加入合適的著色劑、矯味劑、穩定劑和增稠劑來制備。口服使用的水性混懸劑可通過將細小分散的活性組分與粘性物質一道分散在水中,所述粘性物質如為天然合成膠、樹脂、甲基纖維素、羧甲基纖維素和其他藥劑領域已知的懸浮劑。
藥物組合物可為單位劑量形式。在這些形式中,將所述組合物分成含適量活性組分的單位劑量。該單位劑量形式可為包裝制劑,包裝中包括分隔量的制劑,例如盒裝片劑、膠囊劑和在管形瓶或安瓿中的粉劑。單位劑量形式還可為膠囊劑、扁囊劑或片劑或其可為適當數量的任何這些包裝形式。
本發明的藥物組合物,其活性成分可僅為本發明的化合物,也可與其他抗腫瘤化合物組合作為活性成分。
在治療腫瘤的過程中,可采用本發明的藥物組合物與其他抗腫瘤藥進行聯合治療。例如,與用于醫學腫瘤學的抗增殖/抗腫瘤藥、細胞生長抑制劑、抗入侵藥物、生長因子功能抑制劑、抗血管生成劑、血管損傷劑等聯用。
在治療腫瘤時,可通過同時、序貫或單獨給予各種治療成分可實現這種聯合治療。此類組合產品應用有效劑量范圍內的本發明化合物和準許劑量范圍內的其他藥學活性劑。
下面結合實施例對本發明的具體實施方式做進一步的描述,并不因此將本發明限制在所述的實施例范圍之中。
實施例1化合物2的合成
取isobenzofuran-1(3H)-one 10.0g,對羥基苯乙酸11.3g溶于60ml DMF中,加熱至120℃。再緩慢加入30ml含有8.0g甲醇鈉的甲醇溶液,完畢后減壓除去甲醇,回流過夜。加入200ml冰水,濃鹽酸調節pH到12,過濾得到粗品,70%的乙醇重結晶,得到白色固體A,收率60%。1H-NMR(DMSO-d6,400MHz),δH 12.65(2H,s),7.92(1H,dd,J=7.6,1.2Hz),7.63(1H,dd,J=7.1,1.1Hz),7.56(1H,td,J=7.1,1.1Hz),7.44(1H,td,J=7.6,1.2Hz),7.17(2H,d,J=8.6Hz),6.90(2H,d,J=8.6Hz),5.43(2H,s),3.48(2H,s);13C-NMR(DMSO-d6,100MHz)δC173.4,168.6,157.6,138.9,132.6,132.6,130.9,130.9,129.9,128.3,128.1,127.8,114.9,114.9,68.0,40.0。
取白色固體A(即2-(11-oxo-6,11-dihydrodibenzo[b,e]oxepin-2-yl)acetic acid)5.0g,85%的磷酸1.8mmol,乙酰氯1.2eq,溶于50ml甲苯中,加熱至100℃反應10小時。再加入活性炭500mg,在100℃攪拌1小時,過濾冷卻至室溫,得到粗品,乙酸乙酯-正己烷重結晶得淺黃色固體B,收率70%。1H-NMR(CDCl3,400MHz),δH8.12(1H,d,J=2.3Hz),7.88(1H,dd,J=7.6,1.1Hz),7.55(1H,td,J=7.4,1.3Hz),7.46(1H,td,J=7.6,1.1Hz),7.42(1H,dd,J=8.4,2.3Hz),7.36(1H,d,J=7.4Hz),7.03(1H,d,J=8.4Hz),5.18(2H,s),3.67(2H,s);13C-NMR(CDCl3,100MHz),δC 190.9,177.2,160.6,140.4,136.4,135.5,132.8,132.6,129.5,129.3,127.8,127.1,125.2,121.2,73.6,39.9。
取8.0g 1,3-二溴丙烷,三苯基膦10.40g溶于50ml甲苯中,混合物加熱至130℃反應2小時,冷卻至室溫,過濾得到白色粉末三苯基膦溴丙烷,產率90%。
取三苯基膦溴丙烷10.0g,二甲胺水溶液20ml,溶于100ml乙醇中,70℃反應6小時,加壓除去溶劑,乙醇重結晶得到白色晶體C,收率99%。
取白色晶體C 10.0g溶于20ml THF中,在-10℃下緩慢加入95mmol(eq)的正丁基鋰,攪拌1小時后緩慢加入含有4.0g淺黃色固體B的THF溶液,反應液加熱回流10小時,冷卻至室溫,減壓除去溶劑,加入20ml水,乙醚萃取3次,水層酸化至pH=2,乙酸乙酯萃取。水層調pH=7,除去溶劑得到產物D,收率30%。1H-NMR(DMSO-d6,400MHz),δH12.3(1H,br s),7.25-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.1,2.2Hz),6.78(1H,d,J=8.1Hz),5.65(1H,t,J=7.2Hz),5.12(2H,br s),3.56(2H,s),3.25(2H,t,J=7.7Hz),2.79(2H,q,J=7.4Hz),2.72(6H,br s);13C-NMR(CDCl3,100MHz),δC 173.4,154.3,145.0,141.4,133.9,132.3,131.1,129.6,128.2,128.2,127.7,127.3,126.4,123.3,119.7,69.9,55.9,42.2,42.2,40.0,24.9
取上步產物D 10.0g,溶于30ml甲醇中,加入3滴濃硫酸,室溫反應過夜,加壓蒸干溶劑,硅膠柱純化,乙酸乙酯-石油醚系統純化的油狀產物E,收率72%。1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.67(1H,s),3.45(3H,s),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz)δC 172.2,154.6,145.6,139.7,133.7,132.0,130.7,130.0,129.1,127.5,127.5,126.3,125.7,123.9,119.8,70.4,59.5,53.4,52.0,45.4,40.2,28.1.MS(ESI),m/z352.2[M+H]+.
取上步產物E 5.0g,溶于20ml水合肼中,室溫攪拌10小時,乙酸乙酯萃取,飽和食鹽水洗滌3次,無水硫酸鈉干燥,過濾,除去溶劑,硅膠柱層析純化,得終產物(即化合物2),收率77%。1H-NMR(CDCl3,400MHz),δH 7.51-7.16(4H,m),7.08(1H,d,J=2.2Hz),7.02(1H,dd,J=8.3,2.2Hz,1H),6.88(1H,d,J=8.3Hz),5.96(1H,t,J=7.2Hz),5.43(2H,brs,),3.63(2H,s),3.41(2H,m),2.34(2H,t,J=7.2Hz),2.23(6H,br s);13C-NMR(CDCl3,100MHz),δC 172.4,152.1,144.8,139.7,133.7,130.9,130.6,129.9,129.6,128.7,128.7,127.9,127.2,126.7,117.8,71.2,58.2,45.6,39.9,28.8.MS(ESI),m/z352.7[M+H]+。
實施例2化合物1的合成
化合物1的合成方法同實施例2,唯一的不同為將水合肼改為氨水,即得化合物1。收率:64%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.58(2H,br s),3.45(2H,s),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz),δC173.6,154.8,145.5,139.6,133.6,132.2,130.9,130.0,129.2,127.6,127.5,126.6,126.3,124.2,120.3,70.4,59.4,45.4,45.4,42.5,28.3.MS(ESI),m/z337.8[M+H]+。
實施例3化合物8的合成
取中間體酸(即實施例1的產物D)5.0g,DIEA(5eq)溶于30ml DMF中,室溫反應20min,再加入環丙胺1.0eq,HBTU 1.0eq,室溫反應8小時。加入純凈水50ml,乙酸乙酯萃取兩次,水洗3次,飽和食鹽水洗3次,無水硫酸鈉干燥,加壓除去溶劑得粗品,石油醚-乙酸乙酯硅膠柱層析的純品。收率,66%。1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.71(1H,t,J=7.2Hz),5.60(2H,br s),3.44(2H,s),2.64(1H,m),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),0.71(2H,q,J=7.0Hz),0.39(2H,q,J=7.0Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.4,45.4,45.4,42.8,28.2,22.7,6.6,6.6.MS(ESI),m/z377.2[M+H]+。
實施例4化合物3的合成
化合物3的合成方法同實施例3,唯一的不同為將環丙胺改為間甲胺,即得化合物3。收率:65%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.49(2H,br s),3.48(2H,s),3.18(2H,q,J=6.9Hz),2.73(3H,d,J=4.8Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.4,45.4,45.4,42.8,28.3,26.5.MS(ESI),m/z351.2[M+H]+。
實施例5化合物4的合成
化合物4的合成方法同實施例3,唯一的不同為將環丙胺改為間乙胺,即得化合物4。收率:70%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.22(2H,m),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.04(3H,t,J=7.2Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.3,45.4,45.3,42.9,34.5,28.2,14.8.MS(ESI),m/z365.1[M+H]+。
實施例6化合物5的合成
化合物5的合成方法同實施例3,唯一的不同為將環丙胺改為間二甲胺,即得化合物5。收率:55%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.18(2H,q,J=6.9Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.38(2H,m),1.24(2H,m),0.86(3H,t,J=7.2Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.4,45.4,45.4,42.9,39.4,31.6,28.2,20.0,13.8.MS(ESI),m/z393.4[M+H]+。
實施例7化合物6的合成
化合物6的合成方法同實施例3,唯一的不同為將環丙胺改為丙胺,即得化合物6。收率:85%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.15(2H,q,J=6.6Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.43(2H,m),0.83(3H,t,J=7.4Hz).13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.3,45.3,43.0,41.5,38.6,28.1,22.6,11.2.MS(ESI),m/z379.4[M+H]+。
實施例8化合物7的合成
化合物7的合成方法同實施例3,唯一的不同為將環丙胺改為異丙胺,即得化合物7。收率:70%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.37(2H,q,J=7.1Hz),3.29(2H,q,J=7.1Hz),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.11(6H,t,J=7.1Hz);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,59.3,45.3,43.0,41.5,38.6,28.1,22.6,22.6.MS(ESI),m/z379.2[M+H]+。
實施例9化合物9的合成
化合物9的合成方法同實施例3,唯一的不同為將環丙胺改為羥乙胺,即得化合物9。收率:67%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.58(2H,m),3.45(2H,s),3.30(2H,m),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s);13C-NMR(CDCl3,100MHz)δC 170.4,154.7,145.6,139.7,133.6,132.3,130.8,130.0,129.2,127.6,127.5,126.7,126.3,124.1,120.2,70.4,60.9,59.6,45.4,45.4,43.0,41.4,28.5.MS(ESI),m/z381.3[M+H]+。
實施例10化合物10的合成
化合物10的合成方法同實施例3,唯一的不同為將環丙胺改為3-甲氧基丙胺,即得化合物10。收率:82%,1H-NMR(CDCl3,400MHz),δH 7.20-7.40(4H,m),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.83(1H,d,J=8.3Hz),5.72(1H,t,J=7.2Hz),5.43(2H,br s),3.45(2H,s),3.30(4H,m),3.02(3H,s),2.56(2H,m),2.45(2H,t,J=7.2Hz),2.22(6H,br s),1.66(2H,m).13C-NMR(CDCl3,100MHz)δC 171.3,154.7,145.7,139.6,133.5,132.4,130.9,130.2,129.1,127.5,127.5,126.6,126.3,124.2,120.1,71.8,70.4,59.4,58.5,45.4,45.4,43.0,38.5,28.8,28.1.MS(ESI),m/z409.3[M+H]+。
實施例11化合物11的合成
化合物11的合成方法同實施例3,唯一的不同為將環丙胺改為2-氨基噻唑,即得化合物11。收率:80%,1H-NMR(DMSO-d6,400MHz),δH 12.28(1H,s),7.46(1H,d,J=3.6Hz),7.20-7.40(4H,m),7.19(1H,d,J=3.6Hz),7.06(1H,d,J=2.2Hz),7.04(1H,dd,J=8.3,2.2Hz),6.78(1H,d,J=8.3Hz),5.68(1H,t,J=7.2Hz),5.15(2H,br s),3.68(2H,s),2.46(2H,m),2.35(2H,t,J=7.2Hz),2.07(6H,br s);13C-NMR(DMSO-d6,100MHz)δC 169.8,158.5,154.4,145.7,139.1,138.1,134.0,132.2,131.8,130.5,129.6,128.2,127.9,127.3,126.3,123.8,119.7,113.9,69.9,59.1,45.3,45.3,41.1,27.9.MS(ESI),m/z422.1[M+H]+。
實施例12~15
化合物12~15的合成方法同實施例3,唯一不同為將環丙胺改為異丁胺、正丁胺、甲基乙基胺或二乙胺,得到化合物12~15。
試驗例1化合物1~11的激酶抑制活性及體外細胞增殖抑制測試
本實驗的目的是檢測本發明的化合物對體外PARP酶抑制活性以及MDA-MB-436細胞增殖抑制活性,采用的方法分別為商業試劑盒及MTT法。
parp1酶活實驗是利用Trevigen’s PARP1試劑盒(Trevigen,cat.no.4676-096-K)測得,通過GraphPad Prism5software(San Diego,CA,USA)計算得到IC50,結果見表1。
MDA-MB-436細胞增殖抑制實驗結果見表1。
表1
表中,a:PARP-1的IC50是由4點劑量響應曲線計算的估算值。
b:MDA-MB-436的IC50是由4點劑量響應曲線計算的估算值。
c:n.d即not determinded,表示無法確定。
實驗結果表明,化合物1~7、9、12、13對parp1具有較強的抑制活性以及在體外對MDA-MB-436細胞具有較強的增殖抑制活性。其中,Iniparib、Olaparib為現有的PARP抑制劑。
試驗例2化合物2對多種腫瘤細胞的抗增值試驗
采用化合物2做多種腫瘤細胞(包括MDA-MB-231,MDA-MB-468and MDA-MB-436細胞系)的抗增值活性試驗,其結果見圖1。其中,圖1A為用不同濃度的化合物2處理MCF-7,MDA-MB-231,MDA-MB-436,MDA-MB-468細胞系,通過MTT測定了細胞的存活率。圖1B為Hoechst 33258熒光染色檢測細胞DNA損傷(Scale bar=200μm)。圖1C為用不同濃度的化合物2處理MDA-MB-436細胞系24小時,通過流式分析測定的凋亡率。圖1D為化合物2處理MDA-MB-436細胞,利用蛋白質印記法測定了Bax,Bcl-2,Caspase-3,PARP1和PAR的表達量。圖1E為用化合物2處理MDA-MB-436細胞,測定其抑制細胞轉移的能力(Scale bar=100μm)。
從圖1中可以明顯看出,化合物2對多種腫瘤細胞具有顯著的抗增殖活性,包括MDA-MB-231,MDA-MB-468and MDA-MB-436等細胞系。特別是在BRCA1突變的MDA-MB-436細胞系中,其IC50為5.14μM.(Figure 4A)。通過Hoechst 33258染色在熒光顯微鏡下細胞發生凋亡的形態學特征(Figure 4B)。并且細胞凋亡率呈現顯著的劑量依賴(Figure 4C)。
BRCA1功能缺失會導致基因組不穩定,從而導致通過同源重組完成的DNA修復缺陷。因此,BRCA1缺失的細胞DNA損傷率高,并且對parp依賴的DNA修復更加敏感。因此,我們檢測了化合物2對parp1的抑制活性以及下游底物蛋白,如PAR。我們發現,化合物2可顯著抑制PARP1的活性,并且PAR的表達量也顯著降低(Figure 4D)。隨后,我們檢測了化合物2誘導細胞死亡的凋亡標志物。發現,化合物2上調Bax的表達以及下調Bcl-2的表達,caspase-3的活性形式是顯著增加。此外,我們還發現,化合物2可抑制乳腺癌細胞遷移(Figure 4E)。這些結果表明,化合物2可以誘導細胞凋亡,通過抑制PARP1抑制BRCA1基因突變的乳腺癌細胞的遷移。
試驗例3化合物2的體內抗腫瘤實驗
本實驗的目的是檢測發明化合物的體內抗腫瘤效果。本實驗采用模型,測試發明化合物2的體內抗腫瘤活性。所用細胞株為MDA-MB-436。
1.實驗方法
將培養好的MDA-MB-436細胞經消化胰酶消化后,再用PBS液清洗2次,然后用1%臺盼蘭染色,于細胞記數板上進行細胞記數,并調節活細胞濃度至1×107/ml,在無菌條件下施行裸鼠右側胸壁第二乳墊,脂肪層下接種0.2ml/只,共接種3只,術后荷瘤裸鼠繼續飼養于SPF環境中,待腫瘤長至0.8cm3時,行裸鼠間原位移植。在無菌條件下取出乳腺癌組織,剪切成1mm3左右的小塊,分別移植于24只裸鼠右側胸壁的第二乳墊脂肪層下,4d后即可見腫瘤生長,成瘤率100%。裸鼠分組(n=6)并注射給藥。對照組注射PBS,藥物對照組注射Iniparib,用量為100mg/kg/d,實驗組分別注射低劑量的化合物2,用量為12.5mg/kg/d),高劑量的化合物2,用量為25mg/kg/d,
腫瘤生長曲線:從第一次注射開始每隔2天,觀察一次荷瘤裸鼠的全身情況及各組腫瘤生長情況,在無菌的條件下測量各組荷瘤裸鼠移植瘤的直徑,并連續觀察紀錄。以時間為橫坐標,腫瘤的體積為縱坐標,腫瘤體積計算公式為:V=1/2*ab2,a為腫瘤長徑,b為短徑,并據此繪制移植瘤生長曲線。
抑瘤率:注射藥物28天后,脫頸處死各組裸鼠,用動物天平稱取裸鼠體重,在分離各組瘤體,稱取瘤重,計算抑瘤率。
觀察指標:每3天觀測一次裸鼠,觀察有無腹瀉,抽搐,皮疹,體重明顯減輕等反應。2實驗數據
實驗測定的生長曲線如圖2A~圖2D,實驗完畢后解剖所得腫瘤照片如圖2E和圖2F。其中,圖2A為小鼠腫瘤相對體積(n=6)注射PBS,Iniparib(100mg/kg/d),化合物2,低劑量(12.5mg/kg/d),高劑量(25mg/kg/d),**,P<0.01。圖2B為小鼠相對瘤重,注射PBS,Iniparib(100mg/kg/d),化合物2,低劑量(12.5mg/kg/d)高劑量(25mg/kg/d)*,P<0.05;***,P<0.001與對照組相比。圖2C為.治療期間的小鼠體重,**,P<0.01;與對照組相比。圖2D為不同組小鼠的肝、脾、腎*,P<0.05;**,P<0.01;與對照組相比。圖2E為Ki-67和PAR的免疫組化分析(Scalebar=200μm)。圖2F為腫瘤離體組織的PARP,PAR和Caspase-3的蛋白印記分析。
實驗結果表明,化合物2對MDA-MB-436細胞具有明顯的體內生長抑制活性,在每天12.5mg/kg及以上劑量下,可以明顯抑制腫瘤生長或完全消退腫瘤。給藥過程中未發現裸鼠出現體重降低、皮疹、腹瀉等不良反應,表明在測試劑量下,化合物2在給藥劑量范圍內毒性低。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 苯并雙(噻二唑)衍生物、包含其的油墨、以及使用了其的有機電子裝置的制造方法
- 具有含硫取代基的殺有害生物活性雜環衍生物的制造方法與工藝
- 具有含硫取代基的殺有害生物活性雜環衍生物的制造方法與工藝
- 一種苯并噻咯類衍生物及其制備方法和有機發光器件與制造工藝
- 苯并噻吩并咔唑類衍生物及其制備方法和有機發光器件與制造工藝
- 苯并二氮雜*衍生物的2-萘磺酸鹽及晶型和它們的制備方法與制造工藝
- 靛紅雜合喹唑啉類化合物合成的靛紅衍生物及其在制備抗腫瘤藥物中的應用的制造方法與工藝
- 一種雜環衍生物及使用該雜環衍生物的有機發光器件的制造方法與工藝
- 一種二氰基甲烯基苯并四氫呋喃衍生物及其制備方法與制造工藝
- 一種含氮雜環衍生物及使用該含氮雜環衍生物的有機發光器件的制造方法與工藝
- 苯并二氮雜*衍生物的2-萘磺酸鹽及晶型和它們的制備方法與制造工藝
- 一種二氰基甲烯基苯并四氫呋喃衍生物及其制備方法與制造工藝
- 苯并吲唑類衍生物及其制備方法、應用與制造工藝
- 一種二苯并環庚烷衍生物的制備方法與制造工藝
- Atropurpuran的苯并咪唑基和嗎啉基衍生物組合物用于防治胰腺纖維化的制造方法與工藝
- 一種苯并咪唑衍生物鎘配合物及其制備方法和應用
- 一種咪唑并吡啶巰乙酸類衍生物及其制備方法與應用
- Artalbic acid的O?(苯并咪唑基)乙基與O?(二羥乙胺基)乙基衍生物的組合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基與二羥乙胺基衍生物的組合物用于制備抗骨質疏松藥物的制作方法
- 阿掏比克酸苯并咪唑基與二羥乙胺基衍生物的組合物用于制備防治胰腺纖維化藥物的制作方法