鹵素或含氮基團取代的聯芐類似物及其制備方法和用途與流程
本發明屬于藥物合成領域,涉及一類聯芐類似物及其制備方法和用途,具體涉及一類鹵素或含氮基團取代的聯芐類似物及其制備方法和用途。
背景技術:
惡性腫瘤常稱癌癥(cancer),是嚴重威脅人類健康的常見病、多發病。應用傳統細胞毒類抗腫瘤藥(antineoplastic drugs)或抗癌藥(anticancer drugs)進行化療在腫瘤的綜合治療(synthetic therapy)中占有重要地位。但化療藥物對占惡性腫瘤90%以上的實體瘤的治療目前仍未能達到滿意的療效,腫瘤化療存在兩大主要障礙:包括藥物的毒性反應和耐藥性的產生。細胞毒類抗腫瘤藥由于對腫瘤細胞缺乏足夠的選擇性,在殺傷腫瘤細胞的同時,對正常的組織細胞也產生不同程度的損傷作用,毒性反應成為腫瘤化療時藥物用量受限的關鍵因素。化療過程中腫瘤細胞對藥物產生不敏感現象即耐藥性是腫瘤化療失敗的重要原因,亦是腫瘤化療急需解決的難題。
近十幾年來,隨著腫瘤分子生物學和腫瘤藥理學的理論和生物技術的不斷發展,抗腫瘤藥正從傳統的細胞毒作用向針對機智的多環節作用的方向發展,其中新生血管生成抑制劑受到了越來越多的關注。
血管生成(angiogenesis)是指從已有的毛細血管或毛細血管后靜脈發展而形成新的血管。腫瘤生長依賴于血管生成的概念始于20世紀70年代初,但其重要性并未受到關注。近二十年,由于發現了血管生成因子對血管生成的作用,以及血管生成對腫瘤生長和侵入轉移、特別對腫瘤早期發生的重要影響,血管生成成為近年來腫瘤研究的熱點之一,為腫瘤治療開辟了一個新的思路。
聯芐類化合物是指兩個苯甲基單元通過甲基的碳碳單鍵相連而成的一類化合物。天然聯芐具有抗腫瘤、植物生長調節、抑制血管新生等作用,但他們的結構相對單一、主要在于苯環上羥基與甲氧基取代位置和數量的變化。且這些天然聯芐在植物中含量少,不容易大量獲得。
技術實現要素:
本發明的目的在于提供一系列鹵素或含氮基團取代的聯芐類似物,并對這些化合物進行抗腫瘤活性測試和抑制血管新生測試,從而提供具有藥用價值的聯芐類似物新化學實體。
本發明的目的可通過以下措施來達到;
一種鹵素或含氮基團取代的聯芐類似物或其在藥學上可接受的鹽、水合物,所述的鹵素或含氮基團取代的聯芐類似物如式I所示:
其中,A為如式II所示的鹵素或含氮基團取代的苯基或含1-2個N的五元或六元不飽和含氮雜環;
B、C、D可分別獨立的選自氫、鹵素、除甲氧基之外的烷氧基、硝基、胺基或取代胺基,但B、C、D不能同時為氫。
優選的,B、C、D可分別獨立的選自氫、鹵素或硝基,但B、C、D不能同時為氫。
進一步優選的,B、C分別獨立的選自氫、鹵素,D選自氫、鹵素或硝基,但B、C、D不能同時為氫。
本發明所述的鹵素為氟、氯、溴;所述的除甲氧基之外的烷氧基為C2-C5的烷氧基;所述的含1-2個N的五元或六元不飽和含氮雜環為2-吡啶基、3-吡啶基、4-吡啶基、嘧啶-5-基、嘧啶-2-基、吡咯-2-基、吡咯-3-基、吡唑-3-基、吡唑-4-基、咪唑-2-基、咪唑-4-基。
最優選的,本發明所述的鹵素或含氮基團取代的聯芐類似物自:4-羥基-3,5-二甲氧基-3’,4’-二氯聯芐、4’-硝基-4-羥基-3,5-二甲氧基聯芐、4-羥基-3,5-二甲氧基-4’-氯聯芐、4-羥基-3,5-二甲氧基-2’-氯聯芐、4-羥基-3,5-二甲氧基-4’-溴聯芐、4-羥基-4’-氟-3,5-二甲氧基聯芐、2,6-二甲氧基-4-(2-(吡啶-2-基)乙基)苯酚、2,6-二甲氧基-4-(2-(吡啶-4-基)乙基)苯酚、4-羥基-2’-氟-3,5-二甲氧基聯芐、4-羥基-3,5-二甲氧基-3’-氟聯芐、4-羥基-3,5-二甲氧基-2’,4’-二氯聯芐、4-羥基-3,5-二甲氧基-2’,3’-二氯聯芐。
本發明的另一目的在于提供一種鹵素或含氮基團取代的聯芐類似物的制備方法。
所述的鹵素或含氮基團取代的聯芐類似物的制備方法,包括:以4-羥基-3,5-二甲氧基苯甲醛為原料,與乙酸酐反應生成3,5-二甲氧基-4-乙酰氧基苯甲醛,通過硼氫化鈉還原得到3,5-二甲氧基-4-乙酰氧基苯甲醇,再與二氯亞砜反應得到3,5-二甲氧基-4-乙酰氧基苯甲醇,進一步與三苯基膦反應生成(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷;(3,5-二甲氧基-4-乙酰 氧基芐基)-三苯基氯化磷與A-CHO反應生成相應的順反混合的二苯乙烯再經甲醇鈉酯交換得最后經氫氣鈀碳催化得目標化合物
其中,A為如式II所示的鹵素或含氮基團取代的苯基或含1-2個N的五元或六元不飽和含氮雜環;
B、C、D可分別獨立的選自氫、鹵素、除甲氧基之外的烷氧基、硝基、胺基或取代胺基,但B、C、D不能同時為氫。
優選的,B、C、D可分別獨立的選自氫、鹵素或硝基,但B、C、D不能同時為氫。
進一步優選的,B、C分別獨立的選自氫、鹵素,D選自氫、鹵素或硝基,但B、C、D不能同時為氫。
本發明中,所述的A-CHO具體為3,4-二氯苯甲醛、對硝基苯甲醛、對氯苯甲醛、鄰氯苯甲醛、對溴苯甲醛、對氟苯甲醛、2-吡啶甲醛、4-吡啶甲醛、2-氟苯甲醛、3-氟苯甲醛、2,4- 二氯苯甲醛、2,3-二氯苯甲醛。
(1)、(2)、(3)三步反應產率高,達88.7%~99.8%,只需通過萃取將所得產物直接用于下一步反應,第(4)步反應產物會在體系中大量沉淀,通過抽濾得到白色純品(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷。第(5)步反應為關鍵步驟,投料順序和投料比例對反應成敗至關重要,將(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷與A-CHO先溶于無水溶劑,充分攪拌,冰浴下緩慢加入縛酸劑氫化鈉,其中(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷、A-CHO和氫化鈉的摩爾比為1∶1∶1~1∶0.8∶1.2,(5)、(6)步反應只需通過萃取直接進入下一步反應,不需要柱色譜純化;第(7)步反應結束通過硅膠柱色譜分離純化得目標產物,洗脫劑為石油醚-乙酸乙酯(10∶1~5∶1V/V)混合溶劑或石油醚-丙酮(20∶1~3∶1)混合溶劑。
發明人對合成的鹵素或含氮基團取代的聯芐類似物進行體外五種癌細胞:結腸癌HCT116細胞、肺癌A549細胞、肝癌HepG2細胞、乳腺癌MDA-MB-231細胞和胃癌MKN-45細胞的細胞毒實驗測試,表現出不同程度的抗癌作用,其中,對結腸癌HCT116具有最強的抗腫瘤活性,表明這類化合物具有顯著的抑制腫瘤細胞增殖的作用。因此,本發明的另一個目的是提供所述的鹵素或含氮基團取代的聯芐類似物或其在藥學上可接受的鹽、水合物在制備治療腫瘤藥物中的應用。所述的腫瘤為結腸癌、肺癌、肝癌、乳腺癌、胃癌。
發明人對合成的鹵素或含氮基團取代的聯芐類似物以斑馬魚為模型,對它們進行抑制血管新生活性評價,體內斑馬魚血管新生抑制實驗初篩結果表明這些化合物對斑馬魚血管新生表現出不同程度的抑制作用。進一步通過濃度梯度考察活性化合物對斑馬魚血管新生抑制作用,發現對斑馬魚呈現出濃度依賴性的血管新生抑制活性。這對于治療與抗血管新生相關的腫瘤生長與轉移、糖尿病視網膜疾病等具有重要意義。因此,本發明的另一個目的是提供所述的鹵素或含氮基團取代的聯芐類似物或其在藥學上可接受的鹽、水合物在制備抑制血管新生藥物方面的應用。所述的抑制血管新生藥物方面的應用包括在制備治療抗血管新生相關的腫瘤生長與轉移、糖尿病視網膜藥物方面的應用。
本發明的有益效果:
本發明通過簡單高效制備方法合成了一系列鹵素或含氮基團取代的聯芐類似物,以4-羥基-3,5-二甲氧基苯甲醛,先后經過乙酸酐乙酰基保護、硼氫化鈉還原、二氯亞砜氯代和三苯基膦回流制得(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷關鍵中間體。關鍵中間體依次與不同取代的苯甲醛發生維悌希反應、甲醇鈉酯交換反應、氫氣鈀碳還原;最終合成一系列目標產物。反應條件溫和,容易重復,中間步驟不需要純化處理,簡化了實驗過程,且總收率在32%~51%。這些合成的聯芐類似物在抗腫瘤和抑制血管新生方面表現出不同程度的活性。 是潛在的抗腫瘤、治療抗血管新生相關的腫瘤生長與轉移、糖尿病視網膜等疾病的新化學實體,對于相關疾病新藥的開發具有重要意義。
附圖說明
圖1為本發明血管轉基因熒光斑馬魚受精后48小時體節間血管(ISVs)模型;其中,箭頭指示體節間血管。圖1A和圖1A’為陰性對照試驗;圖1B-圖1D和圖1B’-圖1D’是濃度分別為1.25μM,2.5μM和5μM的化合物4加藥組斑馬魚試驗;圖1E和圖1E’是陽性對照試驗(2.5μM SU5416)。
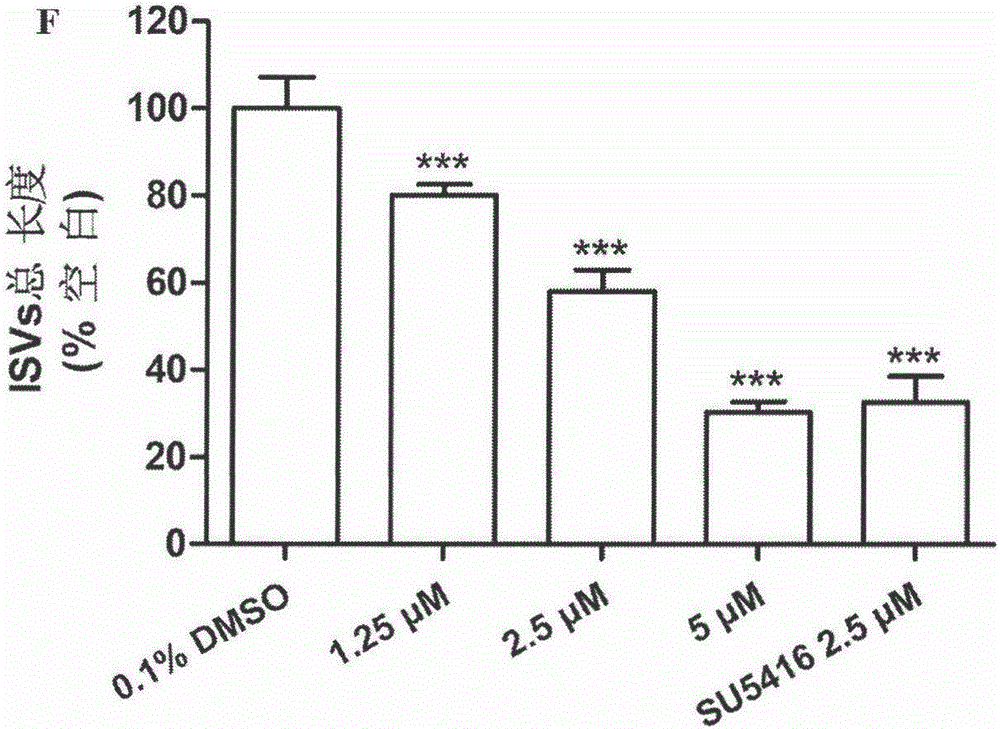
圖2是斑馬魚受精后24小時加入化合物4作用24小時,即受精后48小時斑馬魚的體節間血管(ISVs)總長度統計圖。
具體實施方式
結合具體實施例對本發明的技術方案做進一步詳細的說明,但本發明并不限于以下實施例。
實施例1 4-羥基-3,5-二甲氧基-3’,4’-二氯聯芐(化合物1)的制備
a、3,5-二甲氧基-4-乙酰氧基苯甲醛
在100mL單頸瓶中加入丁香醛(4-羥基-3,5-二甲氧基苯甲醛)粉末8.33g(0.046mol),二氯甲烷10mL,催化計量DMAP(4-二甲氨基吡啶),攪拌使充分溶解。緩慢滴加乙酸酐4.75mL(0.051mol),室溫反應1h。加水洗去DMAP和乙酸酐,共洗3次,每次用水10mL。無水Na2SO4干燥有機層,濃縮回收二氯甲烷,得白色結晶10.19g,產率99.8%。經鑒定為3,5-二甲氧基-4-乙酰氧基苯甲醛。
ESI-MS m/z:225[M+H]+,相對分子質量224。
1H-NMR(300MHz,CDCl3)δ9.93(1H,s,-CHO),7.17(2H,s,H-2,H-6),3.92(6H,s,2×-OCH3),2.38(3H,s,-COCHx)。
b、3,5-二甲氧基-4-乙酰氧基苯甲醇
在100mL單頸瓶中加入3,5-二甲氧基-4-乙酰氧基苯甲醛8.6g(0.038mol),乙醇10mL,充分攪拌。冰浴下緩慢加入NaBH4530mg(0.014mol),室溫反應0.33h。滴加5%HCl調pH 至弱酸,減壓回收溶劑,殘渣加10mL水混懸,用二氯甲烷萃取3次,每次10mL。合并有機層,無水Na2SO4干燥,濃縮二氯甲烷,得白色固體8.34g,產率96.5%。經鑒定為3,5-二甲氧基-4-乙酰氧基苯甲醇。
ESI-MS m/z:227[M+H]+,相對分子質量226。
1H-NMR(300MHz,CDCl3)δ6.66(1H,s,H-2,H-6),4.69(2H,s,Ar-CH2-),3.84(6H,s,2×-OCH3),2.36(3H,s,-COCH3)。
c、3,5-二甲氧基-4-乙酰氧基苯甲基氯
在100mL單頸瓶中加入3,5-二甲氧基-4-乙酰氧基苯甲醇8.3g(0.037mol),二氯甲烷10mL,攪拌溶解。冰鹽浴下加入Et3N 6.2mL(0.043mol),SOCl23mL(0.043mol),室溫反應3h。加入冰水停止反應,用二氯甲烷萃取3次,每次10mL。合并有機層,無水Na2SO4干燥,減壓蒸干二氯甲烷,得淡黃色油狀物7.96g,產率88.7%。經鑒定為3,5-二甲氧基-4-乙酰氧基苯甲基氯。
ESI-MS m/z:245[M-H]-,247[M+H]+,相對分子質量246。
1H-NMR(300MHz,CDCl3)δ6.85(1H,s,H-2,H-6),4.73(2H,s,Ar-CH2-),3.76(6H,s,2×-OCH3),2.24(3H,s,-COCH3)。
d、(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷
在100mL單頸瓶中加入3,5-二甲氧基-4-乙酰氧基苯甲基氯7.8g(0.032mol),三苯基膦8.4g(0.032mol),甲苯5mL,攪拌溶解,加熱回流10h。反應液降至室溫,減壓抽濾,用甲苯洗滌濾餅,干燥得白色粉末14.63g,產率90.2%。經鑒定為(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷。
ESI-MS m/z:541.2[M+Cl]-,相對分子質量506.5。
1H-NMR(300MHz,DMSO-d6)δ7.92(3H,m,ArH),7.76(6H,dt,J=3.6Hz,8.1Hz,ArH),7.69(6H,m,ArH),6.30(2H,d,J=2.4Hz,H-2,H-6),5.09(2H,J=15Hz,Ar-CH2-),3.37(6H,s,2×-OCH3),2.21(3H,s,×-OCH3)。
e、4-羥基-3,5-二甲氧基-3’,4’-二氯聯芐(化合物1)
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),3,4-二氯苯甲醛69mg(0.395mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點即順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-3’,4’-二氯二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-3’,4’-二氯二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶乙酸乙酯=10∶1),得淡黃色結晶67mg,產率51%。經鑒定為4-羥基-3,5-二甲氧基-3’,4’-二氯聯芐。
ESI-MS m/z:349.0[M+Na]+,325.1[M-H]-,相對分子質量為326。
1H-NMR(300MHz,CDCl3)δ7.34(1H,d,J=8.1Hz,H-5’),7.27(1H,brs,H-2’),6.98(1H,dd,J=1.8Hz,8.1Hz,H-6’),6.35(1H,s,H-2,H-6),5.41(1H,brs,ArOH),3.86(6H,s,2×-OCH3),2.84(4H,m,ArCH2CH2Ar)。
實施例2 4’-硝基-4-羥基-3,5-二甲氧基聯芐(化合物2)的制備
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),對硝基苯甲醛60mg(0.397mmol)。氬氣保護下加入無水THF 3mL,攪拌呈淡黃色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點即順反異構二苯乙烯((Z/E)-4’-硝基-3,5-二甲氧基-4-乙酰氧基二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點 完全消失,證實脫乙酰基完全((Z/E)-4’-硝基-4-羥基-3,5-二甲氧基二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶乙酸乙酯=5∶1),得亮黃色油狀物46mg,產率39%。經鑒定為4’-硝基-4-羥基-3,5-二甲氧基聯芐。
ESI-MS m/z:304.1[M+H]+,相對分子質量303。
1H-NMR(300MHz,CDCl3)δ8.15(2H,d,J=8.7Hz,H-3’,H-5’),7.31(2H,d,J=8.7Hz,H-2’,H-6’),6.34(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.0(4H,m,ArCH2CH2Ar)。
實施例3 4-羥基-3,5-二甲氧基-4’-氯聯芐(化合物3)的制備
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),對氯苯甲醛56mg(0.398mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-4’-氯二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-4’-氯二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶乙酸乙酯=10∶1),得黃色固體41mg,產率36%。經鑒定為4-羥基-3,5-二甲氧基-4’-氯聯芐。
ESI-MS m/z:315.1[M+Na]+,相對分子質量為292。
1H-NMR(300MHz,CDCl3)δ7.25(2H,d,J=8.4Hz,H-3’,H-5’),7.09(2H,d,J=8.1Hz,H-2’,H-6’),6.34(2H,s,H-2,H-6),5.43(1H,s,ArOH),3.85(6H,s,2×-OCH3),2.86(4H,m,-CH2CH2-)。
實施例4 4-羥基-3,5-二甲氧基-2’-氯聯芐(化合物4)的制備
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),鄰氯苯甲醛50μL(0.427mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’-氯二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-2’-氯二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶乙酸乙酯=10∶1),得白色固體54mg,產率47%。經鑒定為4-羥基-3,5-二甲氧基-2’-氯聯芐。
ESI-MS m/z:315.1[M+Na]+,291.1[M-H]-,相對分子質量為292。
1H-NMR(300MHz,CDCl3)δ7.38(1H,m,H-3’),7.18-7.11(3H,m,H-4’,5’,6’),6.39(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.87(6H,s,2×-OCH3),3.05-2.84(4H,m,-CH2CH2-)。
實施例5 4-羥基-3,5-二甲氧基-4’-溴聯芐(化合物5)的制備
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),對溴苯甲醛73mg(0.395mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-4’-溴二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點 完全消失,證實脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-4’-溴二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶乙酸乙酯=10∶1),得黃色油狀物42mg,產率32%。經鑒定為4-羥基-3,5-二甲氧基-4’-溴聯芐。
ESI-MS m/z:359.0[M+Na]+,335.1[M-H]-,相對分子質量為336。
1H-NMR(300MHz,CDCl3)δ7.42-7.02(4H,m,ArH),6.37(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.86(6H,s,2×-OCH3),2.95-2.85(4H,m,-CH2CH2-)。
實施例6 4-羥基-4’-氟-3,5-二甲氧基聯芐(化合物6)的制備
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),對氟苯甲醛43μL(0.402mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-4’-氟二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-4’-氟二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶乙酸乙酯=10∶1),得淡黃色固體38mg,產率35%。經鑒定為4-羥基-4’-氟-3,5-二甲氧基聯芐。
ESI-MS m/z:299.1[M+Na]+,275.1[M-H]-,相對分子質量為276。
1H-NMR(300MHz,CDCl3)δ7.10(2H,m,H-3’,H-5’),6.97(2H,m,H-2’,H-6’),6.34(2H,s,H-2,H-6),5.41(1H,s,ArOH),3.85(6H,s,2×-OCH3),2.85(4H,m,-CH2CH2-)。
實施例7 4-羥基-2’-氟-3,5-二甲氧基聯芐(化合物7)
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),鄰氟苯甲醛45μL(0.428mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’-氟二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-2’-氟二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶丙酮=20∶1),得黃色油狀物36mg,產率33%。經鑒定為4-羥基-2’-氟-3,5-二甲氧基聯芐。
ESI-MS m/z:299.1[M+Na]+,相對分子質量為276。
1H-NMR(300MHz,CDCl3)δ7.21-7.01(4H,m,ArH),6.38(2H,s,H-2,H-6),5.40(1H,brs,ArOH),3.86(6H,s,2×-OCH3),2.86(4H,m,-CH2CH2-)。
實施例8 4-羥基-3,5-二甲氧基-3’-氟聯芐(化合物8)
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),間氟苯甲醛45μL(0.424mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-3’-氟二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實反應脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-3’-氟二苯乙烯)。滴加10%稀 鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶丙酮=20∶1),得黃色油狀物44mg,產率37%。經鑒定為4-羥基-3,5-二甲氧基-3’-氟聯芐。
ESI-MS m/z:299.1[M+Na]+,相對分子質量為276。
1H-NMR(300MHz,CDCl3)δ7.23(1H,m,H-2’),6.95-6.88(3H,m,ArH),6.36(2H,s,H-2,H-6),5.43(1H,brs,ArOH),3.86(6H,s,2×-OCH3),2.87(4H,m,-CH2CH2-)。
實施例9 4-羥基-3,5-二甲氧基-2’,4’-二氯聯芐(化合物9)
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),2,4-二氯苯甲醛69mg(0.394mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’,4’-二氯二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實反應脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-2’,4’-二氯二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶丙酮=10∶1),得黃色油狀物52mg,產率40%。經鑒定為4-羥基-3,5-二甲氧基-2’,4’-二氯聯芐。
ESI-MS m/z:349.0[M+Na]+,相對分子質量為326。
1H-NMR(300MHz,CDCl3)δ7.39(1H,d,J=2.1Hz,H-3’),7.14(1H,dd,J=1.8,8.1Hz,H-5’),7.03(1H,d,J=8.4Hz,H-6’),6.36(2H,s,H-2,H-6),5.43(1H,brs,ArOH),3.86(6H,s,2×-OCH3),3.01-2.80(4H,m,-CH2CH2-)。
實施例10 4-羥基-3,5-二甲氧基-2’,3’-二氯聯芐(化合物10)
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),2,3-二氯苯甲醛69mg(0.394mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構二苯乙烯((Z/E)-3,5-二甲氧基-4-乙酰氧基-2’,3’-二氯二苯乙烯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實反應脫乙酰基完全((Z/E)-4-羥基-3,5-二甲氧基-2’,3’-二氯二苯乙烯)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶丙酮=10∶1),得黃色油狀物42mg,產率32%。經鑒定為4-羥基-3,5-二甲氧基-2’,3’-二氯聯芐。
ESI-MS m/z:349.0[M+Na]+,325.2[M-H]-,相對分子質量為326。
1H-NMR(300MHz,CDCl3)δ7.34(1H,dd,J=1.5,7.8Hz,H-4’),7.10(1H,t,J=7.8Hz,H-5’),7.01(1H,dd,J=1.5,7.5Hz,H-6’),5.43(1H,brs,ArOH),3.87(6H,s,2×-OCH3),3.08-2.83(4H,m,-CH2CH2-)。
實施例11 2,6-二甲氧基-4-(2-(吡啶-2-基)乙基)苯酚(化合物11)
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),2-吡啶甲醛40μL(0.420mmol)。氬氣保護下加入無水THF 3mL,攪拌呈亮黃色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成一個新的斑點推測為順反異構((Z/E)-2,6-二甲氧基-4-(2-(吡啶-2-基)乙烯基)苯基乙酸酯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點 完全消失,證實反應脫乙酰基完全((Z/E)-2,6-二甲氧基-4-(2-(吡啶-2-基)乙烯基)苯酚)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶丙酮=3∶1),得黃色結晶38mg,產率37%。經鑒定為2,6-二甲氧基-4-(2-(吡啶-2-基)乙基)苯酚。
ESI-MS m/z:282.1[M+Na]+,258.1[M-H]-,相對分子質量為259。
1H-NMR(300MHz,CDCl3)δ8.57(1H,d,J=3.9Hz,H-3’),7.57(1H,dt,J=1.8,7.8Hz,H-4’),7.12(1H,m,H-5’),7.08(1H,d,J=7.8Hz,H-6’),6.39(2H,s,H-3,H-5),5.89(1H,brs,ArOH),3.81(6H,s,2×-OCH3),3.10-2.94(4H,m,-CH2CH2-)。
實施例12 2,6-二甲氧基-4-(2-(吡啶-4-基)乙基)苯酚(化合物12)
在50mL單頸瓶中加入(3,5-二甲氧基-4-乙酰氧基芐基)-三苯基氯化磷200mg(0.395mmol),4-吡啶甲醛40μL(0.425mmol)。氬氣保護下加入無水THF 3mL,攪拌呈白色混懸液。冰鹽浴下緩慢加入NaH 11mg(0.458mmol),攪拌過夜。TLC監測生成兩個新的斑點推測為順反異構點((Z/E)-2,6-二甲氧基-4-(2-(吡啶-4-基)乙烯基)苯基乙酸酯),濃硫酸乙醇顯色呈淡紫色,證明反應成功。加冰水終止反應,二氯甲烷萃取三次,有機層無水Na2SO4干燥,減壓濃縮備用。
將濃縮物加5mL甲醇溶解,加入適量CH3ONa,反應0.5h。點板監測反應,順反異構點完全消失,證實反應脫乙酰基完全((Z/E)-2,6-二甲氧基-4-(2-(吡啶-4-基)乙烯基)苯酚)。滴加10%稀鹽酸調pH值至中性,減壓濃縮溶劑。加入10mL水混懸,用二氯甲烷萃取三次,每次10mL,有機層無水Na2SO4干燥,減壓濃縮。將濃縮物用5mL甲醇溶解,加入催化劑量10%Pd/C,通入氫氣反應48h。抽濾得澄清甲醇溶液,即含目標化合物的粗品。用硅膠柱色譜純化(石油醚∶丙酮=3∶1),得白色固體36mg,產率35%。經鑒定為2,6-二甲氧基-4-(2-(吡啶-4-基)乙基)苯酚。
ESI-MS m/z:282.1[M+Na]+,相對分子質量為259。
1H-NMR(300MHz,CDCl3)δ8.50(2H,d,J=5.7Hz,H-3’,5’),7.09(2H,d,J=4.0Hz,H-2’,6’),6.33(2H,s,H-3,H-5),5.31(1H,brs,ArOH),3.84(6H,s,2×-OCH3),2.89(4H,m,-CH2CH2-)。
表1 本發明制得的鹵素或含氮基團取代的聯芐類似物
實施例14 本發明化合物體外抑制腫瘤細胞增殖實驗(MTT法)
1、材料
(1)細胞株及試劑
人肺癌細胞株A549、人肝癌細胞株HepG2、人胃癌細胞株MKN-45、乳腺癌細胞株MDA-MB-231、結腸癌細胞株HCT116均購自中國科學院上海生命科學研究院細胞庫;
高糖DMEM培養液購自美國Gibco公司;
胎牛血清購自杭州四季青公司;
胰酶細胞消化液購自上海碧云天生物技術有限公司;
噻唑藍(MTT)購自sigma公司。
PBS緩沖鹽溶液:磷酸二氫鉀(KH2PO4)0.27g,磷酸氫二鈉(Na2HPO4)1.42g,氯化鈉(NaCl)8g,氯化鉀(KCl)0.2g,加去離子水約800mL充分攪拌溶解,然后加入濃鹽酸調pH至7.4, 最后定容到1L,高溫高壓滅菌后置于4攝氏度冰箱保存待用。
(2)受試藥品溶液
化合物1-12用DMSO溶解配制成25mM的溶液,用高糖DMEM培養液稀釋為250μM的母液,在此基礎上依次用高糖DMEM培養液稀釋得25,5,1,0.2,0.04μM的受試藥品溶液,4℃保存備用。
(3)細胞培養
人肺癌細胞A549、人肝癌細胞HepG2、人胃癌細胞MKN-45、乳腺癌細胞MDA-MB-231、結腸癌細胞HCT116在含10%胎牛血清的DMEM培養液中,37℃、5%CO2保持飽和濕度培養,2-3天傳代一次。
分別取對數生長期癌細胞一瓶,加入胰酶細胞消化液消化,使貼壁細胞脫落,配制成濃度為5×104個/mL的細胞懸液,96孔細胞培養板中每孔加入100μL細胞懸液(每孔5×103個細胞),板置于37℃,5%CO2培養箱中培養24小時。棄去培養液,PBS緩沖鹽溶液清洗2次,每孔加入100μL相應的含不同濃度受試藥品溶液的高糖DMEM培養液;同時設立陰性對照組(只加高糖DMEM培養液)。
96孔板置于37℃,5%CO2培養箱中培養72小時,每孔加入10μL MTT溶液(5mg/mL),在培養箱繼續培養4小時。棄去培養基,每孔加入150μL DMSO溶解,搖床10分鐘輕輕混勻;每孔的吸光度(OD值)用1500酶標儀在490nm波長下進行檢測,計算抑制率和IC50值。
應用GraphPad Prism 5軟件計算IC50。
表2 化合物1-12對癌細胞的細胞毒活力測試結果
由表2可知,化合物1-12對結腸癌HCT116細胞、肺癌A549細胞、肝癌HepG2細胞、乳腺癌MDA-MB-231細胞和胃癌MKN-45細胞表現出不同程度的抗癌作用,其中,對結腸癌HCT116具有最強的抗腫瘤活性。結果表明本發明聯芐類似物具有顯著的抑制腫瘤細胞增殖的作用。
實施例15 以轉基因斑馬魚為模型評價本發明化合物抗血管新生抑制活性
1、材料
(1)實驗動物
轉基因斑馬魚Tg(flil:EGFP)購自南京大學模式生物,成魚魚齡:6個月-9個月;飼養光照條件:14h/10h明暗交替,溫度28.5℃,pH 7.2-7.6。實驗全程均使用轉基因斑馬魚幼體胚胎(受精后24小時)作為實驗對象。
(2)藥物與試劑
本發明合成的鹵素或含氮基團取代的聯芐類似物化合物1-12;
紅十字小丑鹽(Instant Ocean salt):Instant Ocean公司;
二甲基亞砜(DMSO):美國Sigma-Aldrich公司;
鏈霉蛋白酶(Pronase):美國Sigma公司;
三卡因(Tricaine):美國Sigma公司;
VEGFR(血管內皮細胞生長因子受體)特異性血管新生抑制劑SU5416:美國Sigma公司。
(3)主要實驗儀器
倒置熒光顯微鏡(Olympus IX71);
超純水機(Milli-Q PLUS 185);
恒溫培養箱:上海知楚儀器有限公司;
體視顯微鏡(Nikon SMZ745)。
2、主要實驗溶液和藥物的配制
卵水(Egg water)配制方法:1L去離子水加入0.2g紅十字小丑鹽(Instant Ocean salt);
0.1%DMSO溶液(陰性對照)配制:使用時用卵水配制成濃度為0.1%的工作液,現配 現用;
VEGFR(血管內皮細胞生長因子受體)特異性血管新生抑制劑SU5416作為本組實驗陽性對照藥,使用時用0.1%DMSO溶液配制成實驗所需的濃度,陽性對照藥濃度為2.5μM;12個鹵素或含氮基團取代的聯芐類似物的血管新生抑制初篩濃度均為50μM,有抑制活性的化合物再用0.1%DMSO溶液稀釋尋找化合物抑制血管新生的最低有效劑量范圍。
3、方法
本實驗材料為發育正常的受精后24小時齡轉基因斑馬魚(可在血管內皮細胞特異性表達綠色熒光蛋白)。使用鏈霉蛋白酶(0.5mg/mL)將其卵膜消化除去得到脫去卵膜的斑馬魚胚胎。以20個斑馬魚胚胎/孔將斑馬魚隨機分配至含1mL培養液的24孔板內,并立即向孔內培養液中加入對應濃度的化合物溶液,陰性對照組加入等量的0.1%DMSO溶液,加藥完畢后將24孔板轉移至28.5℃恒溫生物培養箱中。繼續培養24小時(即胚胎發育至受精后48小時),取出孔板,使用倒置熒光顯微鏡觀察,拍攝血管熒光照片并對斑馬魚血管新生的情況進行統計。
體內斑馬魚血管新生抑制實驗結果見表3,大部分化合物表現出不同程度的血管新生抑制活性,其中化合物4的抑制斑馬魚血管新生活性強于陽性藥。
以化合物4為代表,圖1表示斑馬魚體節間血管生成的抑制率隨著化合物4濃度的上升而呈現梯度增加,即化合物對斑馬魚血管新生呈現出濃度依賴性的抑制活性。圖2為化合物4對斑馬魚體節間血管生長抑制作用的統計圖。
表3 化合物1-12對斑馬魚血管新生抑制活性
- 還沒有人留言評論。精彩留言會獲得點贊!