TCO綴合物和治療劑遞送方法與流程
本申請要求2014年11月21日提交的美國臨時申請號62/083,022、2014年6月18日提交的62/013,994以及2014年3月14日提交的61/953,294的優先權,各個以全部內容并入文本用于所有目的。
關于對在聯邦政府資助的研發下作出的發明的權利的聲明
不適用。
技術實現要素:
在第一實施方式中,本發明提供一種固相支持體組合物,具有生物相容的固相支持體,至少一結合(劑),以及具有約1至約10個連接原子的連接體,每個結合劑共價連接至生物相容的固相支持體。
在另一實施方式中,本發明提供一種生物活性組合物,其具有治療或診斷劑,可以是反式環辛烯或四嗪的結合劑,以及具有約1至約10個連接原子的連接體,結合劑共價連接至治療或診斷劑。
在另一實施方式中,本發明提供
的組合物。
在另一實施方式中,本發明提供用于選擇性遞送有效量的治療劑或診斷劑至患者的目標器官或組織的第一位置的方法。該方法包括在患者的目標器官或組織的第一位置植入本發明的固相支持體組合物,其中,固相支持體組合物包含第一結合劑。該方法還包括給患者施用本發明的生物活性組合物,其中,生物活性組合物包括第二結合劑,且其中第一和第二結合劑在接觸后彼此結合,從而選擇性遞送有效量的治療或診斷劑至患者的目標器官或組織的第一位置。
在另一實施方式中,本發明提供用于選擇性遞送有效量的治療劑或診斷劑至患者的目標器官或組織的第一位置的方法。該方法包括在患者的目標器官或組織的第一位置植入固相支持體組合物,其具有生物相容的固相支持體和連接到固相支持體的第一結合劑。該方法還包括給患者施用生物活性組合物,其具有治療劑或診斷劑、與第一結合劑互補的第二結合劑以及連接治療或診斷劑和第二結合劑的可釋放的連接體,以使第一和第二結合劑在接觸后彼此結合。該方法還包括釋放診斷或治療劑,從而將診斷或治療劑遞送至目標器官或組織的第一位置。
附圖簡述
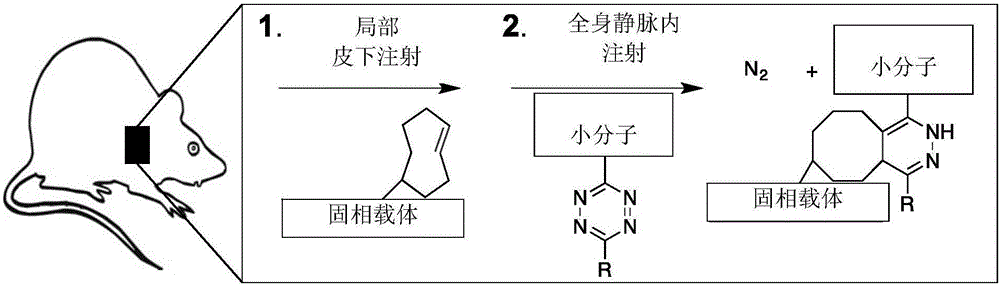
圖1A和圖1B示出本發明的方法,涉及初始注射共價連接到TCO的生物材料。隨后是全身注射偶聯到四嗪部分的治療劑。當兩個實體接近時,它們共價反應連接治療至固相載體,從而定位治療劑。含有放射性111Im(銦)的Tz放射性探針2用作分子探針。
圖2示出體外研究,預稱重的凝膠盤(實驗或對照)與Tz放射性探針2的溶液混合10分鐘或14小時。在10分鐘的暴露期,樣本TCO-凝膠1結合1.78nmoles/g Tz放射性探針2,而對照凝膠維持0.75nmoles/g Tz放射性探針2。在14小時內,連接到對照凝膠的Tz放射性探針的量保持不變為0.86nmoles/g。TCO-凝膠1結合2.78nmoles/g。誤差條代表三個重復實驗的平均值的標準偏差。星號表示經配對t檢驗的統計顯著性(p值<0.05)。
圖3A和圖3B顯示Tz放射性探針2的生物分布結果。a、接受皮下TCO-凝膠1(平均0.23克[0.10-0.29克])和對照凝膠(平均0.23克[0.16-0.25克])的小鼠,經尾靜脈注射Tz放射性探針2(平均44μCi[39.2–48.4μCi])。b、在特定時間點TCO-凝膠1與對照凝膠的平均差異。柱體表示在特定時間點(1,4,24,48小時)每個器官的%ID/克。誤差條表示三個重復實驗的平均值的標準偏差。星號表示經配對t檢驗的統計顯著性(p值<0.05)。
圖4示出接受皮下TCO-凝膠1(170mg,綠色區域)和對照凝膠(230mg,黃色區域)的小鼠。植入后3小時,經尾靜脈注射遞送Tz放射性探針2(1.05mCi)并通過SPECT在4,24,48小時成像。
圖5示出固相支持體組合物TZ-Me-凝膠的合成制備。
圖6示出通過非可釋放的連接體與TCO連接的熒光素與四嗪固相支持體之間的反應。R代表完整二乙胺熒光素部分,仍共價地連接到固相支持體。
圖7示出通過可釋放的連接體與TCO連接的熒光素與四嗪固相支持體之間的反應。如所預期的,熒光素二乙胺(MW:418.40)和二氧化碳從固相支持體釋放。
圖8提供除去凝膠后的上清液樣品,用于推斷未反應的TCO-X-熒光素的量及TCO-X-熒光素和TZ-Me-凝膠之間的反應動力學。簡言之,方法涉及:形成和稱重Tz-凝膠,加入1毫升含預定濃度的TCO-X-熒光素(10,20,50納摩爾)的PBS鹽水,將板置于孔板混合器(速度200)預定量的時間(10,50,100,180分鐘),在特定時間點轉移上清液至另一板,和通過IVIS光譜儀檢測殘留在上清液中熒光的量(490nm激發熒光素,515nm發射)。
圖9示出基于實驗值的熒光團量和相關的輻射(熒光)的標準曲線擬合,其中利用已知納摩爾量的TCO-NR-熒光素,用IVIS光譜儀檢測的輻射(490nm激發熒光素,515nm發射)。
圖10示出當和TZ-Me-凝膠在37℃混合0至70分鐘時,上清液中TCO-NR-熒光素隨時間減少。
圖11示出結合到凝膠的TCO-NR-熒光素和上清液中檢測到的未結合的量的理論相互性。
圖12示出根據圖10的數據,結合到TZ-Me-凝膠的TCO-NR-熒光素分子的計算量。基于給出的數據,當TZ-Me-凝膠盤與50納摩爾TCO-NR-熒光素混合時,70分鐘后,每1毫克TZ-Me-凝膠有0.66nmolesTCO-NR-熒光素反應(0.66nmoles/mg)。即使只在70分鐘后,是如圖2所示用TCO-凝膠14小時后獲得的量(2.78nmoles/g)的約200倍。
圖13顯示在37℃孵育預定量時間(0-3天)后TZ-Me-凝膠的活性。在規定的孵育后,加入50納摩爾TCO-NR-熒光素激發50毫克凝膠90分鐘,依照前圖計算結合量。
圖14示出將TCO-R-熒光素加到TZ-Me-凝膠多個時間點后未結合分子的預期與觀察量。考慮到TCO-R-熒光素和TCO-NR熒光素之間的相似之處,預計反應模式類似。假設是上清液中回收熒光的量的差異是由于二乙基熒光素的釋放,如圖7所示。
圖15示出將特定量TCO-R-熒光素(10、20、50納摩爾)加入TZ-Me-凝膠后釋放的熒光素的總量。此外,采用LC-MS分析在時間點60分鐘上清液中化合物的具體特性并顯示存在熒光素二胺(分子量418.40)為主要化合物,還證實存在TCO-R-熒光素與TZ-Me-凝膠的捕獲和釋放反應的消除產物。
圖16A和圖16B示出使用捕獲和釋放連接體的本發明的方法。簡要地說,該方法首先包括含點擊化學載體改性的固體載體的定位,在該特定實例中為四嗪。接著氨基甲酸酯(綠色)和反式環辛烯基團改性的載物暴露于材料(無論體外或體內)。兩試劑原位反應,根據四嗪取代基,載物或從固體載體釋放或固定到固體載體。
圖17示出暴露于不同的四嗪修飾的瓊脂糖珠后TCO-R-羅丹明的釋放動力學。簡言之,如圖16所示在X和Y具有雜環取代基的瓊脂糖1a,導致緩慢釋放(紅線),大概是由于吸電子基團引起的衰減。另一方面,在X上具有芳族取代基和在Y上具有烷基取代基導致羅丹明二乙胺的快速釋放。值是三次實驗的結果。
圖18示出未植入凝膠或植入Tz-凝膠,或僅植入海藻酸,然后注入TCO-R-羅丹明或TCO-NR-羅丹明的小鼠在70分鐘,5小時和3天輻射值。
圖19A和圖19B示出TCO-R-萬古霉素對發光MSSA的MIC(Xen 29)。
圖20示出(R,E)-N-(2-氨乙基)-2-(環辛-4-烯-1-基氧)乙酰胺(5)的NMR。
圖21示出未改性海藻酸凝膠的NMR。
圖22示出TCO-凝膠1的NMR。
圖23示出Tz-Me-凝膠的NMR。
圖24示出“捕獲和釋放”策略,采用生物正交TCO-四嗪化學。
圖25示出180分鐘后熒光素釋放量。計算為結合熒光團的預期值和獲得的實際實驗值之間的差。
圖26示出60分鐘后Tz-Me-凝膠和TCO-R-熒光素混合物上清液中發現的熒光素二胺的峰([M+H]419.1214和[M+MeOH]453.3417),通過液相色譜-質譜確定預期的“捕獲和釋放”產品。
圖27示出暴露于不同四嗪改性的瓊脂糖珠后TCO-R-熒光素([M+H]is 571.21)的釋放動力學。簡言之,如圖16所示在X和Y具有雜環取代基的瓊脂糖1a,導致緩慢釋放(MX53,紅線),大概是由于吸電子基團引起的衰減。另一方面,在X上具有芳族取代基和在Y上具有烷基取代基導致羅丹明二乙胺(MX65,藍線)的快速釋放。左手邊示出通過質譜動態評估釋放。
圖28示出熒光素標記的TCO從四嗪改性瓊脂糖的“捕獲和釋放”的動力學研究設計。
圖29示出通過質譜學方法動態評估TCO-R-羅丹明([M+H]625.31)的釋放。如所預期在473.22發現釋放產物。
圖30示出基于以前的動力學生物分布研究,注射TCO-R-羅丹明后在各時間點比較陰性對照(無凝膠注射)和釋放方案(TZ-Me-凝膠)熒光的增加。柱狀圖推斷8小時內獲得推定總量的數據。在凝膠存在下的曲線下面積反映的劑量幾乎是雙倍的,如“釋放”柱(右)相對陰性對照(左)所示。
圖31示出小鼠的動力學多劑量評估。在各自通過尾靜脈注射50納摩爾TCO-R-羅丹明后對三只小鼠進行評價。示出如上所述處理小鼠的圖像。“陰性對照”對象不接受任何凝膠注射。“釋放”對象在背部皮下接受Tz-Me-凝膠注射。“凝膠對照”對象在背部皮下接受海藻酸凝膠注射。在每次給熒光團后5-10分鐘和110-150分鐘后用IVIS光譜儀評價小鼠。
圖32示出在給每個不同實驗組(陰性對照=無凝膠;實驗組=Tz-Me-凝膠;凝膠對照=未改性海藻酸凝膠)第二、第三和第四次注射50納摩爾TCO-R-羅丹明后5-10分鐘相較于110-150分鐘所觀察到的平均熒光值。實驗組熒光在統計學上高于陰性或凝膠對照組。通過配對t檢驗進行比較(n=3)。
具體實施方式
I.概述
本發明提供用于植入和給藥于對象的組合物,其中組合物使用短連接體,其也可以包括可釋放的組分。可釋放的連接體能將更大量的治療劑或診斷劑給藥至有需要的患者。
II.定義
“治療劑”是指能夠治療和/或減輕病癥或疾病的試劑。代表性的治療劑包括但不限于,紫杉醇,多柔比星,依托泊苷,伊立替康,SN-38,環孢菌素A,鬼臼毒素,卡氮芥,兩性霉素,伊沙匹隆,帕妥匹隆(埃博霉素epothelone類),萬古霉素,雷帕霉素和鉑類藥物。本發明的治療劑還包括前藥形式。
“診斷劑”是指幫助診斷病癥或疾病的試劑。代表性的診斷劑包括成像劑,如順磁性試劑,光學探針和放射性核素。順磁性試劑是在外加場下具有磁性的成像劑。順磁性試劑的例子包括但不限于,鐵顆粒包括納米顆粒。光學探針是可以通過一輻射波長激發并在第二、不同的輻射波長檢測進行檢測的熒光化合物。本發明中有用的光學探針包括但不限于,Cy5.5、Alexa(亞歷克薩)680、Cy5、DiD(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚二碳菁高氯酸鹽)和DiR(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚三碳菁碘化物)。其他光學探針包括量子點。放射性核素是經過放射性衰變的元素。本發明中有用的放射性核素包括但不限于:3H、11C、13N、18F、19F、60Co、64Cu、67Cu、68Ga、82Rb、90Sr、90Y、99Tc、99mTc、111In、123I、124I、125I、129I、131I、137Cs、177Lu、186Re、188Re、211At、Rn、Ra、Th、U、Pu和241Am。
“目標器官或組織”是指作為目標用于遞送治療或診斷劑的器官或組織。作為目標的代表性的器官和組織包括能被化學或生物靶向劑所靶向的那些,以及不能被化學或生物靶向劑靶向的那些器官和組織。代表性的器官或組織包括骨。
“選擇性遞送”是指遞送治療或診斷劑至需要治療的器官或組織部分,而不靶向不需要治療的器官或組織的其他部分。
“移植”是指手術植入到患者體內。
“生物相容的固相支持體”是指能夠植入患者體內且支持結合劑之一,以及結合劑結合后的治療或診斷劑的固相支持體材料。固相支持體與患者身體相容。代表性的生物相容的固相支持體包括但不限于,水凝膠如多糖水凝膠,海藻酸,纖維素,殼聚糖,透明質酸,硫酸軟骨素,肝素等。
“接觸的”或“接觸”是指使兩不同物種接觸以使它們能反應的過程。但應理解,得到的反應產物可以直接產生自添加試劑間的反應或產生自能夠在反應混合物中產生的一個或更多添加試劑的中間體。
“連接體”、“連接的”或“連接”是指連接本發明化合物至靶向特定類型細胞,如癌細胞,其他類型疾病細胞或正常細胞類型的生物材料的化學部分。連接可以通過共價或離子鍵形成。連接可以是兩被連接部分直接連接或間接連接,如通過連接體。本發明中有用的連接體長度至多30個碳原子。優選地,連接體長度是5-15個碳原子。用于將接頭連接到本發明的化合物和生物分子的鍵的類型包括但不限于酰胺,胺,酯,氨基甲酸酯,脲,硫醚,硫代氨基甲酸酯,硫代碳酸鹽和硫脲。本領域技術人員將認識到其它類型的鍵在本發明中是有用的。
“結合劑”是指能在生物環境中與另一結合劑形成共價鍵的任何群組。這通常被稱為生物結合或生物正交化學。代表性的結合劑包括但不限于,胺和活化酯,胺和異氰酸酯,胺和異硫氰酸酯,用于形成二硫化物的硫醇,用于形成烯胺的醛和胺,用于通過施陶丁格連接形成酰胺的疊氮化物,用于經由點擊-化學形成三唑的炔烴和疊氮化物,反式-環辛烯(TCO)和四嗪等等。本發明中有用的結合劑與相應的結合劑具有高反應性,以致迅速反應。
“治療”,“治療的”和“治療方法”是指治療或改善損傷,病理,病癥或癥狀(例如,疼痛)的任何成功的標記,包括任何客觀或主觀的參數,如癥狀的減輕、緩解、減弱或使得患者更可容忍癥狀、損傷、病理或病癥;降低病癥或癥狀頻率或持續時間;或者,在某些情況下,預防癥狀或病癥的發作。癥狀的治療或改善可以基于任何客觀或主觀參數,包括例如身體檢查的結果。
“給藥”是指口服給藥,以栓劑給藥,局部接觸,胃腸外,靜脈內,腹膜內,肌內,病灶內,鼻內或皮下給藥,鞘內給藥,或植入緩釋裝置,如,微型滲透泵,給對象。
“患者”是指需要治療的動物,如哺乳動物,包括但不限于,靈長類(例如人),牛,綿羊,山羊,馬,狗,貓,兔,大鼠,小鼠等。在某些實施方式中,患者是人。
“治療有效量或劑量”或“治療足夠量或劑量”或“有效或足夠量或劑量”是指對被給藥者產生治療效果的劑量。確切的劑量取決于治療目的,由本領域技術人員使用已知的技術(參見,如Lieberman,藥物劑型(Pharmaceutical Dosage Forms)(卷1-3,1992);Lloyd,制藥工藝,科學與技術(The Art,Science and Technology of Pharmaceutical Compounding)(1999);Pickar,劑量計算(Dosage Calculations)(1999);和Remington:藥劑學科學和實踐(The Science and Practice of Pharmacy),第20版本,2003,Gennaro編.,Lippincott,Williams和Wilkins)確定。在致敏細胞中,治療有效劑量通常可以低于非致敏細胞傳統的治療有效劑量。
III.組合物
本發明提供組合物,具有短連接體或可釋放的連接體用于選擇性遞送治療或診斷劑給患者。在一些實施方式中,本發明提供固相支持體組合物,具有生物相容的固相支持體,至少一結合劑,和具有約1-約10個連接原子的連接體,將各結合劑共價連接至生物相容的固相支持體。
任何合適的生物相容的固相支持體可以用于本發明的方法。例如,生物相容的固相支持體可以是水凝膠、交聯聚合物基質、金屬、陶瓷、塑料等等。用于本發明的水凝膠包括但不限于,多糖凝膠,海藻酸,纖維素,透明質酸,殼聚糖,甲殼素,幾丁質,透明質酸,硫酸軟骨素,肝素等。其他糖基生物材料是現有技術已知的,如聚合物先進技術(Polymer Advanced Technology)2014,25,448-460描述的那些。用作生物相容的支持體的聚合物可以包括但不限于:聚磷腈,聚酐,聚縮醛,聚(原酸酯),聚磷酸酯,聚己內酯,聚氨酯,聚乳酸,聚碳酸酯,聚酰胺和聚醚,以及它們的共混物/復合材料/共聚合物。代表性的聚醚包括,但不限于,聚(乙二醇)(PEG),聚丙二醇(PPG),三嵌段聚醚(Pluronic,[PEG]n-[PPG]m-[PEG]n),PEG二丙烯酸酯(PEGDA)和PEG二甲基丙烯酸酯(PEGDMA)。生物相容的固相支持體也可以包括蛋白質和其他聚(氨基酸),例如膠原,明膠,彈性蛋白和彈性蛋白樣多肽,白蛋白,纖維蛋白,聚(γ-谷氨酸),聚(L-賴氨酸),聚(L-谷氨酸),和聚(天冬氨酸)。
在一些實施方式中,固相支持體可以是水凝膠。在一些實施方式中,固相支持體可以是海藻酸(鹽)。在一些實施方式中,固相支持體可以是幾丁質。在一些實施方式中,固相支持體可以是透明質酸。在一些實施方式中,固相支持體可以是殼聚糖。在一些實施方式中,固相支持體可以是瓊脂糖。
任何合適的連接體可以用于本發明以連接結合劑至生物相容的固相支持體或治療或診斷劑。代表性的連接體可以具有約1-約100個連接原子,且可包括乙烯氧基部分,胺,酯,酰胺,酮,脲,氨基甲酸酯和碳酸酯官能團。用于本發明方法的其他連接體可以具有約1-約50個連接原子,或約1-約10個連接原子,或約5-約10個連接原子。代表性的連接體包括但不限于以下示出的那些:
適用于本發明的其他連接體包括:
任何合適的結合劑可以用于本發明的方法。可以在生物偶聯技術“Bioconjugate Techniques”Greg T.Hermanson,1996和ACS化學生物學(ACS Chemical Biology)2014,9,592-605中找到代表性的結合劑。例如,用于本發明方法的結合劑包括但不限于,環辛烯,四嗪,疊氮化物,炔,胺,活化的酯,異氰酸酯,異硫氰酸酯,硫醇,醛,酰胺,等等。在一些實施方式中,結合劑可以是環辛烯,四嗪,疊氮化物或炔。在一些實施方式中,結合劑可以是反式-環辛烯或1,2,4,5-四嗪。在一些實施方式中,結合劑可以是反式-環辛烯。在一些實施方式中,結合劑可以是4-甲基-1,2,4,5-四嗪。
在一些實施方式中,結合劑和連接體共同結構為:
在一些實施方式中,組合物結構為:
本發明還提供通過連接體連接到結合劑的治療劑或診斷劑的組合物。在一些實施方式中,本發明提供生物活性組合物,具有治療或診斷劑、可以是反式-環辛烯或四嗪的結合劑,以及具有約1-約10個連接原子的連接體,共價連接結合劑至治療或診斷劑。
任何治療或診斷劑可以用于本發明的方法。代表性的治療劑包括但不限于,抗生素如萬古霉素,紫杉醇,多柔比星,依托泊苷,伊立替康,SN-38,環孢菌素A,鬼臼毒素,卡氮芥,兩性霉素,伊沙匹隆,帕妥匹隆(埃博霉素epothelone類),雷帕霉素和鉑類藥物。其它治療劑包括多西環素和其它MMP抑制劑。仍然其它治療劑包括達托霉素,左旋多巴,奧司他韋,頭孢氨芐,5-氨基乙酰丙酸,半胱氨酸,制霉菌素,兩性霉素B,氟胞嘧啶,恩曲他濱,甲氧芐啶,磺胺甲惡唑,阿昔洛韋,塞來考昔,尼莫地平,強力霉素,頭孢曲松,等等。在一些實施方式中,治療劑可以是萬古霉素。在其他實施方式中,治療劑可以是達托霉素。在另外的實施方式中,治療劑可以是阿霉素。在另一實施方式中,治療劑可以是環磷酸腺苷(c-AMP)。
代表性的診斷劑包括成像劑,如順磁性試劑,光學探針和放射性核素。順磁性試劑在外加場下具有磁性的成像劑。順磁性試劑的例子包括但不限于,鐵顆粒包括納米顆粒。光學探針是可以通過一輻射波長激發并在第二、不同的輻射波長檢測進行檢測的熒光化合物。本發明中有用的光學探針包括但不限于,Cy5.5、Alexa(亞歷克薩)680、Cy5、DiD(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚二碳菁高氯酸鹽)和DiR(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚三碳菁碘化物)。其他光學探針包括量子點。放射性核素是經過放射性衰變的元素。本發明中有用的放射性核素包括但不限于:3H、11C、13N、18F、19F、60Co、64Cu、67Cu、68Ga、82Rb、90Sr、90Y、99Tc、99mTc、111In、123I、124I、125I、129I、131I、137Cs、177Lu、186Re、188Re、211At、Rn、Ra、Th、U、Pu和241Am。診斷劑也可以包括螯合劑,如1,4,8,11-四氮雜環十二烷-1,4,8,11-四乙酸(TETA)、4,11-雙(羧甲基)-1,4,8,11-四氮雜雙環[6.6.2]十六烷(CB-TE2A)、二乙烯三胺五乙酸(DTPA)和1,4,7,10-四-氮雜環癸烷四乙酸(DOTA)。其它螯合劑在本發明的方法中是有用的。
任何合適的連接體可以用于本發明從而將結合劑連接到生物相容的固相支持體或治療或診斷劑。代表性的連接體可以具有約5到約100個連接原子,且可以包括乙烯-氧部分,胺,酯,酰胺,酮,脲,氨基甲酸酯和碳酸酯官能團。本發明的方法中有用的其它連接體可具有約5至約50個連接原子,或約1至約10個連接原子,或約5至約10個連接原子。代表性的連接體包括但不限于,以下示出的那些:
任何合適的結合劑可以用于本發明的方法。可以在生物偶聯技術“Bioconjugate Techniques”Greg T.Hermanson,1996和ACS化學生物學(ACS Chemical Biology)2014,9,592-605中找到代表性的結合劑。例如,用于本發明方法的結合劑包括但不限于,環辛烯,四嗪,疊氮化物,炔,胺,活化的酯,異氰酸酯,異硫氰酸酯,硫醇,醛,酰胺,等等。在一些實施方式中,結合劑可以是環辛烯,四嗪,疊氮化物或炔。在一些實施方式中,結合劑可以是反式-環辛烯或1,2,4,5-四嗪。在一些實施方式中,結合劑可以是反式-環辛烯。在一些實施方式中,結合劑可以是4-甲基-1,2,4,5-四嗪。
在一些實施方式中,結合劑和連接體共同結構為:
在一些實施方式中,本發明提供的組合物為
IV.選擇性遞送治療或診斷劑的方法
本發明提供通過將本發明的固相支持體組合物植入有此需要的患者,再將本發明的生物活性組合物給藥于患者的選擇性遞送治療或診斷劑的方法。在一些實施方式中,本發明提供選擇性遞送有效量的治療或診斷劑到患者目標器官或組織的第一位置的方法。方法包括將本發明的固相支持體組合物植入患者目標器官或組織的第一位置,其中固相支持體組合物包括第一結合劑。方法還包括將本發明的生物活性組合物給藥于患者,其中,所述生物活性組合物包括第二結合劑,其中第一和第二結合劑在接觸后互相結合,從而選擇性遞送有效量的治療或診斷劑到患者目標器官或組織的第一位置。
使用本發明的方法可以靶向任何合適的器官或組織。代表性的器官或組織包括但不限于,骨,軟骨,韌帶,肌腱,腸,肌肉,神經系統包括腦,脊髓,心臟,神經和其他。例如,當器官是心臟,本發明的方法可用于心臟修復。在一些實施方式中,患者目標器官或組織的第一位置相對于目標器官或組織的其他位置不能有選擇地通過化學或生物靶向劑打靶。在一些實施方式中,目標器官或組織可以是骨。
在以上為固相支持體組合物和生物活性組合物的更多詳述中描述了適于本發明方法的結合劑。在一些實施方式中,第一結合劑可以是反式-環辛烯,第二結合劑可以是四嗪。在一些實施方式中,第二結合劑可以是1,2,4,5-四嗪。在一些實施方式中,第二結合劑可以是4-甲基-1,2,4,5-四嗪。
在一些實施方式中,可植入的組合物具有結構:
在一些實施方式中,可植入的組合物可以是:
和
生物活性組合物可以是:
其中R可以是治療劑或診斷劑。
可以通過本領域技術人員已知的任何方式植入生物相容的固相支持體。
治療或診斷劑可以足以治療患者患有的疾病或病癥的任何合適的量給藥。給藥的劑量、頻率和時間將很大程度上取決于所選擇的治療劑,待治療的病癥的性質,患者的情況包括年齡、體重和其他病癥或疾病癥的存在,待給藥的制劑和主治醫師的判斷。通常,治療劑或診斷劑以每天約2毫克到至多約2000毫克的劑量給藥,然而必然將發生如上所述取決于疾病靶,患者和給藥途徑的變化。本發明的治療或診斷劑可以需要的頻率給藥,包括每小時,每天,每周或每月。本發明的制劑方法中使用的治療或診斷劑以每天約0.0001mg/kg到約1000mg/kg的初始劑量給藥。可以使用每日劑量范圍約0.01mg/kg到約500mg/kg,或約0.1mg/kg到約200mg/kg,或約1mg/kg到約100mg/kg,或約10mg/kg到約50mg/kg。但是,劑量可以根據患者的要求、治療病癥的嚴重性和使用的化合物而變化。例如,考慮特定患者診斷疾病的類型和階段憑經驗確定劑量。施用給患者的劑量,在本發明的上下文中,應隨時間足以產生有益的治療效果,但通常低于沒植入將治療或診斷劑聚集在需要治療器官或組織處的生物相容的固相支持體情況下治療患者所需的劑量。劑量大小還將由伴隨將特定化合物給藥于特定患者的任何不良副作用的存在,性質和程度來確定。特定情況的合適劑量的確定在醫師的技術范圍內。通常,治療開始時用較小的劑量,它小于化合物的最佳劑量。此后,以小的增量增加劑量直至達到情況下的最佳效果。為方便起見,如果需要,總日劑量可以在一天分次部分給藥。劑量可每日給予,或隔天,如由治療醫生所確定的。也可以經更長的時間(周,月或年)定期或連續給予劑量。
在一些實施方式中,目標器官或組織的第一位置的治療或診斷劑的濃度高于患者其他位置的濃度。在一些實施方式中,治療劑是萬古霉素。
在另一實施方式中,本發明提供選擇性遞送有效量的治療或診斷劑到患者目標器官或組織的第一位置的方法。方法包括在患者目標器官或組織的第一位置植入具有生物相容的固相支持體和連接到所述固相支持體的第一結合劑的固相支持體組合物。方法還包括將具有治療或診斷劑,與第一結合劑互補的第二結合劑以及連接治療或診斷劑和第二結合劑的可釋放的連接體的生物活性組合物給藥至患者,以致第一和第二結合劑在接觸后互相結合。方法還包括釋放治療或診斷劑,從而將治療或診斷劑遞送至目標器官或組織的第一位置。
V.制劑
本發明的組合物可以以各種各樣的口服,胃腸外和局部劑型來制備。口服制劑包括適合于患者咽下的片劑,丸劑,粉末,糖衣丸,膠囊,液體,錠劑,扁囊劑,凝膠,糖漿劑,膏劑,混懸劑等。本發明的組合物也可以通過注射,即通過靜脈,肌內,皮內,皮下,十二指腸內,或腹腔內給藥。此外,本文描述的組合物可通過吸入給藥,例如,鼻內給藥。另外,本發明的組合物可以經皮給藥。本發明的組合物還可以通過眼內,陰道內,直腸內給藥,包括栓劑、吹入劑、粉末和氣霧劑(類固醇吸入劑的例子,見Rohatagi,臨床藥理學雜志“J.Clin.Pharmacol.”35:1187-1193,1995;Tjwa,過敏哮喘免疫學年報“Ann.Allergy Asthma Immunol.”75:107-111,1995)。此外,本發明還提供藥物組合物,包括藥學上可接受的載體或賦形劑以及本發明的化合物。
為從本發明化合物制備藥物組合物,藥學上可接受的載體可以是固體或液體。固體形式制劑包括粉劑,片劑,丸劑,膠囊劑,扁囊劑,栓劑和可分散顆粒劑。固體載體可以是一種或多種物質,其也可以作為稀釋劑,調味劑,粘合劑,防腐劑,片劑崩解劑,或包封材料。關于制劑和給藥技術的細節在科學和專利文獻中有很好的描述,見,例如,雷明頓Remington的藥物科學的最新版本,Maack出版公司,伊斯頓PA(“雷明頓的”)。
在粉劑中,載體是細碎的固體,其在含細碎活性組分的混合物中。在片劑中,活性組分以合適的比例與具有所需性能的載體混合并被壓實成所需的形狀和大小。粉劑和片劑優選含有5%或10%到70%的本發明化合物。
合適的固體賦形劑包括但不限于,碳酸鎂;硬脂酸鎂;滑石;果膠;糊精;淀粉;黃蓍膠;低熔點蠟;可可脂;碳水化合物;糖,包括但不限于,乳糖,蔗糖,甘露醇或山梨糖醇;來自玉米,小麥,水稻,馬鈴薯或其他植物的淀粉;纖維素,如甲基纖維素,羥丙基甲基纖維素,或羧甲基纖維素鈉;和樹膠,包括阿拉伯和黃蓍膠;以及蛋白,包括但不限于,明膠和膠原蛋白。如果需要,可加入崩解劑或增溶劑,如交聯聚乙烯吡咯烷酮,瓊脂,海藻酸或其鹽,如海藻酸鈉。
糖衣丸芯具有合適的包衣如濃縮的糖溶液,其還可以含有阿拉伯膠,滑石,聚乙烯吡咯烷酮,卡波姆凝膠,聚乙二醇和/或二氧化鈦,漆液和合適的有機溶劑或溶劑混合物。染料或色素可加入片劑或糖衣丸包衣用于產品識別或表征活性化合物的量(即,劑量)。也可以口服使用本發明的藥物制劑,例如使用如甘油或山梨糖醇的包衣和明膠制成的軟的,密封膠囊,以及明膠制成的推入配合膠囊。推入配合膠囊可以含有本發明的化合物,混合有填充劑或粘合劑如乳糖或淀粉,潤滑劑如滑石粉或硬脂酸鎂,和任選的穩定劑。在軟膠囊中,本發明的化合物可以溶解或懸浮在合適的液體中,例如脂肪油,液體石蠟,或液體聚乙二醇,有或沒有穩定劑。
為了制備栓劑,首先將低熔點蠟,如脂肪酸甘油酯或可可脂的混合物融化,本發明的化合物均勻地分散其中,例如通過攪拌。然后將熔化的均勻混合物倒入適宜大小的模具中,使其冷卻并由此固化。
液體形式制劑包括溶液,懸浮液和乳液,例如,水或水/丙二醇溶液。對于腸胃外注射,液體制劑可以在水性聚乙二醇溶液中配制成溶液。
適合于口服使用的水溶液可通過將本發明的化合物溶解在水中并根據需要加入合適的著色劑,調味劑,穩定劑和增稠劑來制備。適于口服使用的水性混懸液可以通過將細碎的活性組分分散在水中制成,水中含有粘性物質如天然或合成樹膠,樹脂,甲基纖維素,羧甲基纖維素鈉,羥丙基甲基纖維素,海藻酸鈉,聚乙烯吡咯烷酮,黃蓍樹膠和阿拉伯樹膠,和分散或潤濕劑如天然存在的磷脂(例如,卵磷脂),環氧烷烴與脂肪酸的縮合產物(例如,聚乙二醇硬脂酸酯),環氧乙烷與長鏈脂肪醇的縮合產物(例如,十七乙烯氧基十六醇),環氧乙烷與衍生自脂肪酸和己糖醇的偏酯的縮合產物(如,聚氧乙烯山梨糖醇單油酸酯),或環氧乙烷與衍生自脂肪酸和己糖醇酐的偏酯的縮合產物(如,聚氧乙烯山梨糖醇單油酸酯)。水性懸浮液也可含有一種或多種防腐劑例如對羥基苯甲酸乙酯或正丙酯,一種或多種著色劑,一種或多種調味劑和一種或多種甜味劑,如蔗糖,阿司帕坦或糖精。制劑可用滲透度來調節。
還包括固體形式制劑,其在使用之前不久預期轉換為液體形式制劑用于口服給藥。這樣的液體形式包括溶液,懸浮液和乳液。除了活性成分外,這些制劑可以含有著色劑,調味劑,穩定劑,緩沖劑,人工和天然甜味劑,分散劑,增稠劑,增溶劑等。
在另一實施方式中,可以配制本發明的組合物用于腸胃外給藥,如靜脈內(IV)施用或給藥到器官的體腔或內腔。用于給藥的制劑通常包括本發明的組合物溶解于藥學上可接受的載體形成的溶液。能夠被使用的可接受的載體和溶劑有水和林格氏溶液,等滲氯化鈉。此外,無菌固定油可以常規地用作溶劑或懸浮介質。為了這個目的,可以采用任何溫和的固定油,包括合成的單或二甘油酯。此外,脂肪酸如油酸同樣可用于注射劑的制備中。這些溶液是無菌的,并且通常沒有不期望的物質。這些制劑可以通過常規的,眾所周知的滅菌技術進行滅菌。當需要接近生理條件時制劑可含有藥學上可接受的輔助物質,例如pH調節劑和緩沖劑,毒性調節劑,例如,乙酸鈉,氯化鈉,氯化鉀,氯化鈣,乳酸鈉等。本發明的組合物在這些制劑中的濃度可以廣泛變化,并且將根據選擇的特定給藥模式和患者的需求主要基于流體體積,粘度,體重等進行選擇。對于IV給藥,制劑可以是無菌可注射的制劑,如無菌可注射水性或油性懸浮液。這種懸浮液可以根據已知技術用那些適當的分散劑或濕潤劑和懸浮劑配制。無菌可注射制劑還可以是在無毒的胃腸外可接受的稀釋劑或溶劑中的無菌注射溶液或懸浮液,例如1,3-丁二醇溶液。
在另一實施方式中,本發明的組合物的制劑可以通過使用與細胞膜融合或被內吞的脂質體遞送,即通過采用附著到脂質體或直接附著到寡核苷酸的配體,其結合細胞的表面膜蛋白受體導致內吞。通過使用脂質體,特別是在脂質體表面帶有特異于靶細胞的配體的位置,或以其它方式優先定向特定器官,可以將本發明的組合物集中遞送到體內的靶細胞(參見例如,Al-Muhammed,微膠囊雜志“J.Microencapsul.”13:293-306,1996;Chonn,Curr.Opin.Biotechnol.6:698-708,1995;Ostro,Am.J.Hosp.Pharm.46:1576-1587,1989)。
VI.給藥
本發明的組合物可以通過任何合適的方式遞送,包括口服,腸道外和局部方法。經皮給藥方法,通過局部途徑,可以配制成涂藥棒,溶液,懸浮液,乳液,凝膠,霜劑,軟膏劑,糊劑,膠凍劑,涂料,粉末和氣霧劑。
藥物制劑優選為單位劑型。在這樣的形式中,制劑被細分為含有合適量本發明化合物的的單位劑量。單位劑型可以是包裝的制劑,含有離散量制劑的包裝,如包裝的片劑,膠囊和在小瓶或安瓿中的粉劑。此外,單位劑型可以是膠囊,片劑,扁囊劑或錠劑本身,或者它可以是合適數量的這些的包裝形式。
本發明的化合物可以任何合適的量存在,并且可以取決于各種因素,包括但不限于,對象的體重和年齡,疾病的狀態等。本發明化合物的合適劑量范圍包括約0.1毫克至約10,000毫克,或約1mg至約1000mg,或約10毫克到約750毫克,或約25毫克到約500毫克,或約50毫克至約250毫克。本發明的化合物的合適劑量包括約1毫克、5、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900或1000毫克。
本發明的化合物可以任何合適的頻率,間隔和持續時間給藥。例如,本發明的化合物可以一次一小時給藥,或一個小時兩次、三次或更多次,每天一次,或每天兩次、三次或更多次,或每2,3,4,5,6或7天一次,以便提供優選的劑量水平。當本發明的化合物給藥多于每日一次,代表性的間隔包括5,10,15,20,30,45和60分鐘,以及1,2,4,6,8,10,12,16,20和24小時。本發明的化合物可以是施用一次,兩次,或三次或更多次,給藥一小時,1-6小時,1至12小時,1至24小時,6至12小時,12至24小時,一天,1至7天,單周,1至4周,一個月,1至12個月,一年或更多,或者甚至無限期。
本發明的化合物可與另一活性劑共同給藥。共同給藥包括在0.5、1、2、4、6、8、10、12、16、20或24小時彼此施用本發明的化合物和活性劑。共同給藥還包括同時,大致同時(例如,彼此在約1、5、10、15、20或30分鐘內),或以任何次序順序地給藥。此外,本發明的化合物和活性劑可以各自每天一次,或每天兩次、三次或更多次給藥,以提供每天優選劑量水平。
在一些實施方式中,共同給藥可以通過共制劑實現,即制備包括本發明的化合物和活性劑兩者的單一的藥物組合物。在其他實施方式中,本發明的化合物和活性劑可以單獨配制。
本發明的化合物和活性劑可以任何合適的重量比存在于本發明的組合物中,例如從約1:100至約100:1(重量/重量),或約1:50到約50:1,或約1:25至約25:1,或約1:10至約10:1,或約1:5至約5:1(重量/重量)。本發明的化合物和其他活性劑可以任何合適的重量比存在,例如約1:100(重量/重量)、1:50、1:25、1:10、1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、10:1、25:1、50:1或100:1(重量/重量)。本發明的化合物和活性劑的其他劑量和劑量比在本發明的組合物和方法中適用。
VII.實施例
材料除非另有說明,全部試劑和NMR溶劑購自西格瑪-奧爾德里奇Sigma-Aldrich(圣路易斯,密蘇里州)。化合物2獲自虹膜生物技術Iris Biotech(馬克特雷德維茨,德國),而化合物8購自Polypure(奧斯陸,挪威)。DOTA-NHS酯獲自Macrocyclics(達拉斯,德克薩斯州)。硅膠購自Silicycle(魁北克,加拿大),而制備TLC板(20x 20cm;厚度1000μm)購自安萊爾科技Analtech(紐瓦克,DE)。超純海藻酸購自ProNova生物醫學(挪威)。[111In]氯化銦溶液購自珀金埃爾默PerkinElmer(沃爾瑟姆,美國)。稀鹽酸中的[64Cu]氯化銅購自華盛頓大學(圣路易斯,密蘇里州)或采用11MeV西門子RDS 111回旋加速器通過64Ni(p,n)64Cu核反應內部制作并通過陰離子交換色譜法(Biorad AG 1-X8)純化。Dulbecco杜氏磷酸緩沖鹽(DPBS)購自英杰公司(卡爾斯巴德,加利福尼亞最高)。
方法采用瓦里安400MHz VNMRS儀器在CDCL3或[D6]DMSO中進行NMR實驗。在波士頓大學化學儀器中心使用安捷倫離子阱LC/MSD SL得到高分辨率ESI質譜數據,無論是陽性或陰性測定。在有機合成階段,使用裝有沃特斯(Waters)XBridge C18柱(19×250毫米)的安捷倫Agilent 1100系列,應用梯度含0.1%TFA的MeCN和水,進行HPLC純化。
放射化學期間,對于體內分析和純化,使用裝有Jupiter Proteo C-12柱(250×4.6mm,4μm,菲羅門Phenomenex,托倫斯,加利福尼亞州)以及串聯至Bioscan公司的FlowCount光電倍增管(PMT)(Bioscan,華盛頓,哥倫比亞特區)的單波長或二極管陣列UV檢測器(設置220和254nm)的貝克曼-柯爾特系統金(Beckman-Colter System Gold)128色譜系統實施反相HPLC。使用32Karat軟件包(Beckman-Colter)分析數據。流動相由溶劑A:水中0.05%三氟乙酸和溶劑B:100%乙腈構成,1.5毫升/分鐘的流速,注射后2分鐘開始線性梯度,在30分鐘內從9%溶劑B,然后增加至81%,除非另有說明。
使用海藻酸凝膠的體外實驗期間,在裝有沃特斯2487雙吸光度檢測器(220和320毫微米)和Bioscan公司的Flow-count放射性檢測器的沃特斯微風(Waters Breeze)色譜系統上實施分子篩高效液相色譜(HPLC)。采用pH 6.8,0.1M磷酸鈉,以1.0mL/min等度洗脫Phenomenex BioSep SEC-S3000柱(7.8×300mm)。
通過放射-TLC,采用ITLC-SG條(頗爾生命科學Pall Life Sciences,安阿伯,密歇根州)檢測64Cu和111In標記產量,采用0.9%NaCl水溶液中的200mM EDTA洗脫,采用Bioscan 200成像掃描儀(Bioscan,華盛頓哥倫比亞特區)實施。在這些條件下,游離的放射性核素遷移Rf=0.9,而連接到四嗪6的放射性核素保持在原處。
使用Inveon臨床前成像站(西門子醫療解決方案)獲得PET/CT數據。
動物處理根據加利福尼亞大學,戴維斯分校,動物使用和管理委員會認可的方案處理所有動物。
統計學分析組變化描述為平均值±一標準偏差。采用雙尾非配對t檢驗比較單組。P<0.05的組被認為是顯著差異的。微軟Excel版本12.8.9用于所有的統計計算。
實施例1 TCO改性海藻酸1的制備
以下描述TCO改性海藻酸的制備,首先制備TCO-連接體,然后連接至海藻酸。
(R,E)-N-(2-氨乙基)-2-(環辛-4-烯-1-基氧)乙酰胺(5)。化合物3(100mg,0.54mmol)、N-羥基琥珀酰胺(69mg,0.60mmol)和N,N'-二環己基碳二亞胺(123mg,0.60mmol)溶于CH2Cl2(2mL)并在室溫攪拌2小時。通過PVDF膜濾掉沉淀脲。采用CH2Cl2(1mL)清洗膜。將母液加入到攪拌中的乙二胺(324mg,5.4mmol)的CH2Cl2(10mL)溶液。室溫攪拌反應2小時。采用水(2×10mL)洗滌反應混合物。采用硫酸鎂干燥有機層并減壓濃縮。使用4:1的CH2Cl2和MeOH的混合物作為溶劑在制備型硅膠TLC上純化產物。化合物5的產量是60mg(49%)。1H NMR(CDCl3)δ7.08(bs,1H),5.61-5.45(m,2H),3.92(q,J=11.23Hz,2H),3.64(dd,J1=9.91Hz,J2=4.73Hz,1H),3.36(q,J=6.07Hz,2H),2.87(bs,2H),2.31-2.16(m,4H),2.08-2.04(m,1H),1.89-1.49(m,5H)。13C NMR(CDCl3)δ170.34,135.48,131.37,75.80,68.31,41.28,40.05,34.28,32.53,29.74,27.85。HRMS:m/z[M+H]+計算C12H23N2O2227.1754,實測227.1791。
在標準碳化二亞胺化學條件下每克UP MVG海藻酸與176微摩TCO-胺結合,如之前對精氨酸-甘氨酸-天冬氨酸(RGD)和甘氨酸-組氨酸-賴氨酸(GHK)摻合所述。然后通過用含有降低鹽濃度的去離子水透析4天純化海藻酸產物,冷凍并凍干5-10天直到干燥。通過添加DPBS得到2.5%海藻酸溶液,并通過加入鈣制備海藻酸凝膠。通過1H-NMR研究(見圖20-22)證實海藻酸的共價修飾。不添加TCO的相同方案用于構建對照凝膠。用完全相同批次的TCO-凝膠完成體外和體內研究并在同一天使用以最大程度降低負載量或負載效率中的任何變化。
對于體外試驗,800μl 2.5%的海藻酸溶液與200μl過飽和Ca(SO4)2溶液(每毫升雙蒸水(ddH2O)0.21g Ca(SO4)2)。使用三通活塞混合溶液30秒從而獲得2%終濃度海藻酸。混合物在特制塑料模具內的兩塊玻璃板之間膠凝并在室溫孵育20分鐘。總體具有與膠凝作用一致的均勻外觀。用刮鏟拾起盤片,單獨稱重。通常情況下,預制盤重約100毫克,并大致具有如下尺寸:8mm(直徑)和2mm(高度)。
對于體內使用,2.5%的海藻酸凝膠溶液和過飽和硫酸鈣溶液按上述相同的比例迅速混合,并立即以所需量注射到動物。
實施例2制備四嗪-改性診斷劑
如前所述合成Tz輻射探針2(R.Rossin,P.R.Verkerk,S.M.van den Bosch,R.C.Vulders,I.Verel,J.Lub,M.S.Robillard,Angew.Chem.Int.Ed.Engl.2010,49,3375).
實施例3制備(E)-環辛-2-烯基2-氨乙基氨基甲酸酯
根據Versteegen,R.M.等,Angew.Chem.Int.Ed.2013,52,14112-14116中描述的方案制備標題化合物。
實施例4制備可釋放的TCO-改性診斷劑
將熒光素-NHS酯(134mg,0.283mmol)和(Z)-環辛-2-烯基2-氨乙基氨基甲酸酯(60.0mg,0.283mmol)溶于DMF(5mL)。加入三乙胺(77μL,0.566mmol)并在室溫攪拌18h。高真空下蒸發溶劑并將反應混合物重溶于甲醇。通過制備型薄層色譜采用1:9的MeOH:CH2Cl2混合物作為流動相進行純化。產量=90mg(55.6%)。1H NMR(CD3OD,400MHz)δ8.42(s,1H),8.17(d,J=8.2Hz,1H),7.25(d,J=8.2Hz,1H),6.86(s,2H),6.57-6.50(m,4H),5.78(t,J=12.3Hz,1H),5.47(d,J=16.4Hz,1H),5.2(app s,1H),3.61-3.31(m,5H),2.32(bs,1H),1.98-1.88(m,3H),1.83-1.77(m,1H),1.69-1.04(m,5H),0.83-0.75(m,1H).13C NMR(CD3OD,100MHz)δ168.74,161.63,158.85,156.60,154.17,137.94,135.62,132.86,132.70,130.20,125.81,125.31,113.89,111.03,103.81,75.43,41.69,41.21,37.08,36.67,30.14,25.27.HRMS(ESI-MS)m/z:計算C32H30N2O8[M+H+]571.2080;實測571.2025.
實施例5制備TCO-改性診斷劑
將2-((E)-環辛-2-烯基氧)-N-(2-氨乙基)乙酰胺(50.0mg,0.221mmol)和熒光素-NHS酯(105mg,0.221mmol)溶于DMF(5mL)。加入三乙胺(60μL,0.442mmol)并在室溫攪拌18h。高真空下蒸發溶劑并將反應混合物重溶于甲醇。通過制備型薄層色譜采用1:9的MeOH:CH2Cl2混合物作為流動相進行純化。產量=51mg(39.5%)。1H NMR(CD3OD,400MHz)δ8.46(s,1H),8.21(d,J=8.2Hz,1H),7.65(bs,1H),7.28(d,J=8.2Hz,1H),6.69(d,J=2.7Hz,2H),6.58-6.50(m,4H),5.46-5.44(m,2H),3.91(d,J=5.5Hz,2H),3.65-3.59(m,5H),2.67(s,1H),2.32-2.26(m,2H),2.32-2.26(m,2H),2.20-2.13(m,1H),2.08(d,J=4.2Hz,1H),1.96-1.92(m,1H),1.79-1.67(m,3H),1.54-1.46(m,1),1.30-1.25(m,1H),1.23-1.15(m,1H)。13C NMR(CD3OD,100MHz)δ173.60,170.67,168.69,154.21,137.21,137.66,136.66,136.75,132.53,130.30,125.89,125.09,113.84,110.98,103.79,77.56,69.38,41.31,40.75,40.07,39.94,35.50,33.58,30.82,29.05。HRMS(ESI-MS)m/z:計算C33H32N2O8[M+H+]585.2237;實測585.2183.
實施例6制備可釋放的TCO-改性阿莫西林
將(E)-環辛-2-烯基4-硝基苯基碳酸酯(100mg,0.343mmol)和阿莫西林(82.0mg,0.224mmol)溶于DMF(5mL)。加入三乙胺(87μL,0.634mmol)并在室溫攪拌18h。高真空下蒸發溶劑并將反應混合物重溶于甲醇。通過制備型薄層色譜采用1:9的MeOH:CH2Cl2混合物作為流動相進行純化。產量=36mg(31%)。1H NMR(CD3OD,400MHz)δ8.49(bs,1H),7.57(dd,J1=21.8Hz,J2=8.2Hz,1H),7.24(d,J=6.8Hz,2H),6.67(d,J=6.9Hz,2H),5.81–5.76(m,1H),5.51(d,J=16.4Hz,1H),5.29(s,1H),5.17(s,1H),4.92(s,1H),4.33(s,1H),3.62(s,3H),3.38(s,1H),2.50(s,2H),2.38(bs,1H),1.98-1.88(m,3H),1.81-1.75(m,1H),1.64-1.53(m,2H),1.37(s,3H),1.19-1.12(m,4H),0.81-0.78(m,2H)。13C NMR(CD3OD,100MHz)δ170.49,156.77,154.78,132.16,131.87,131.24,130.95,128.60,114.83,72.99,65.73,59.77,59.07,57.53,57.03,56.89,51.93,48.60,35.54,35.31,28.46,27.07,26.76,23.67,23.54。HRMS(ESI-MS)m/z:計算C33H32N2O8[M+MeO-]548.2072;實測548.2042。
實施例7制備TCO-改性阿莫西林
將2-((E)-環辛-2-烯基氧)乙酸(85.0mg,0.461mmol)、N-羥基琥珀酰胺(53.0mg,0.461mmol)和N,N′-二環己基碳二亞胺(95.0mg,0.461mmol)溶于CH2Cl2(5mL)。室溫攪拌18小時。過濾沉淀物并減壓濃縮上清液。加入阿莫西林(160mg,0.438mmol)的DMF(10mL)溶液并在室溫攪拌18h。高真空下蒸發溶劑并將反應混合物重溶于甲醇。通過制備型薄層色譜采用1:9的MeOH:CH2Cl2混合物作為流動相進行純化。產量=20mg(8.6%)。1H NMR(CD3OD,400MHz)δ7.33(t,J=9.5Hz,1H),7.24(t,J=0.6Hz,1H),6.76(d,J=8.2Hz,2H),5.68-5.45(m,4H),4.32(bs,1H),3.99-3.09(m,2H),3.72(s,1H),3.47(bs,1H),2.67(s,6H),2.37-1.96(m,5H),1.85-1.80(m,3H),1.56(s,3H),1.47(s,3H),1.28-1.17(m,3H)。13C NMR(CD3OD,100MHz)δ175.08,172.65,171.82,158.81,131.16,131.07,130.69,130.13,129.98,129.89,116.69,83.19,68.67,66.71,59.88,58.86,57.05,53.13,35.26,35.14,34.26,34.19,27.18,26.75,26.69,26.41,23.44,23.41。HRMS(ESI-MS)m/z:計算C33H32N2O8[M+MeO-]548.2072;實測548.2042。
實施例8制備改性瓊脂糖
NHS活化瓊脂糖吸附柱購自皮爾斯/賽默飛世爾科技公司(Pierce/Thermo Fisher Scientific)(羅克福德,伊利諾斯州)。采用制造商推薦方案將圖16B的1a或1b的胺前體連接至瓊脂糖珠。簡言之,33mg干瓊脂糖與4μmol的1a或1b的胺前體在pH 7.4PBS緩沖液中孵育3小時,溫和地混合。采用PBS洗滌瓊脂糖珠數次。通過監測洗滌獲得的上清液在520nm的吸光度估算結合到柱的1a或1b的量。瓊脂糖上未反應的NHS基團被1M Tris(pH 7.4)封端。
實施例9捕獲和釋放連接體的動力學
用2微摩TCO-R-羅丹明((E)-5-((2-(((環辛-2-烯-1-基氧)羰基)氨基)乙基)氨甲酰基)-2-(3-(二甲基-4-偶氮烯)-6-(二甲基氨基)-3H-氧雜蒽-9-基)苯甲酸)處理采用上述方法的用1a或1b改性的瓊脂糖珠2分鐘。快速離心后收集上清,并將瓊脂糖重懸于水中。在固定時間間隔收集上清并用ESI-MS分析。
在賽默飛世爾科技公司(西棕櫚灘,加利福尼亞州)線性離子阱靜電場軌道阱(LTQ Orbitrap Velos)質譜儀上采用石英毛細管發射器分析樣本。為便于噴霧優化,在質譜分析之前將10%異丙醇加到各樣本。以陽性模式分析釋放產物,羅丹明乙二胺(5-((2-氨乙基)氨甲酰基)-2-(3-(二甲基-4-偶氮烯)-6-(二甲基氨基)-3H-氧雜蒽-9-基)苯甲酸)。單獨合成釋放產物羅丹明乙二胺用于ESI-MS校正(圖SX)。采用校正曲線評估各步驟上清液中羅丹明乙二胺的量。
實施例10制備改性Tz-Me-海藻酸
在標準碳化二亞胺化學條件下每克UP MVG海藻酸與176微摩(4-(6-甲基-1,2,4,5-四嗪-3-基)苯基)甲胺(Tz-Me–胺)結合,如之前對RGD、GHK和TCO胺摻合所述。然后通過用含有降低鹽濃度的去離子水透析4天純化海藻酸產物,冷凍并凍干5-10天直到干燥。通過添加ddH2O得到2.5%海藻酸溶液,并通過加入鈣制備海藻酸凝膠。通過1H-NMR研究(見圖23)證實海藻酸的共價修飾。不添加TCO的相同方案用于構建對照凝膠。用完全相同批次的TCO-凝膠完成體外和體內研究并在同一天使用以最大程度降低負載量或負載效率中的任何變化。
對于體內使用,800微升2.5%的海藻酸溶液和200微升過飽和硫酸鈣溶液(0.21g Ca(SO4)2/ml ddH2O)混合。采用三通活塞混合溶液30秒從而獲得2%海藻酸終濃度。立即以所需量將混合物注射給動物。
實施例11捕獲和釋放連接體的體內評價
IACUC批準后,通過不注射或注射一種海藻酸(對照相對Tz-凝膠)在nu/nu小鼠上實施體內熒光實時生物分布研究。然后對象接受尾靜脈注射TCO-R-F或TCO-NR-F。陰性對照為:1.無凝膠,用TCO-R-F(陰性對照,小鼠1);2.含TCO-R-F的對照海藻酸(凝膠對照,小鼠4)。兩實驗組為Tz-凝膠以及或者TCO-R-F(釋放方案,小鼠2)或TCO-NR-F(固定化方案,小鼠3)。采用IVIS光譜儀(珀金埃爾默,馬薩諸塞州)檢測熒光并記錄輻射。
實施例12可釋放萬古霉素的最小抑制濃度(MIC)
我們在ddH2O中過夜制備含常規海藻酸凝膠或Tz-凝膠的萬古霉素或TCO-R-萬古霉素的連續稀釋液。翌日,將2.2%Mueller Winto肉湯中的發光甲氧西林敏感金黃色葡萄球菌(Staph.Aureus,MSSA,Xen 29,珀金埃爾默,MA)加至混合物。然后將培養板置入培養箱24小時,并使其生長24小時(n=3)。然后采用IVIS光譜儀(珀金埃爾默,馬薩諸塞州)檢測熒光并記錄輻射率。
實施例13制備可釋放的TCO-羅丹明
如Brunet,A.;Aslam,T.;Bradley,M.Bioorg.Med.Chem.Lett.2014,24,3186-3188所描述的合成羅丹明-NHS酯。將羅丹明-NHS酯(50mg,0.095mmol)和(E)-環辛-2-烯基-2-氨乙基氨基甲酸酯(40.0mg,0.190mmol)溶于CH2Cl2(5mL)。加入三乙胺(129μL,0.95mmol)并在室溫攪拌18h。高真空下蒸發溶劑并將反應混合物重溶于甲醇。采用7.5:2.5:90MeOH:Et3N:CH2Cl2混合物作為流動相通過制備型薄層色譜純化。產量=28mg(47%)。1H NMR(CD3OD,400MHz)δ8.54(s,1H),8.04(d,J=8.2Hz,1H),7.34(d,J=8.2Hz,1H),7.23(d,J=9.6Hz,1H),6.99(dd,J1=2.7Hz,J2=9.5Hz,1H),6.89(d,J=2.8Hz,1H),5.85(t,J=13.7Hz,1H),5.55(d,J=16.4Hz,1H),5.26(s,1H),3.67-3.36(m,4H),3.32-3.21(m,9H),2.93-2.77(m,6H),2.49-2.36(m,1H),2.10-1.79(m,5H),1.78-1.41(m,4H),1.39-1.25(m,1H),1.22-1.06(m,10H),0.93-0.79(m,1H).13C NMR(CD3OD,100MHz)δ172.47,169.41,161.68,159.08,158.76,142.06,137.19,132.96,132.70,130.90,129.85,129.63,115.09,114.86,97.53,76.85,75.38,41.96,41.78,41.01,37.18,36.92,30.21,25.35。HRMS(ESI)m/z:計算C36H41N4O6[M+1]+625.3026;實測625.2976.
實施例14制備非-可釋放的TCO-羅丹明
將羅丹明-NHS酯(50mg,0.095mmol)和2-((E)-環辛-4-烯基氧)-N-(2-氨乙基)乙酰胺(43.0mg,0.190mmol)溶于CH2Cl2(5mL)。加入三乙胺(129uL,0.95mmol)并在室溫攪拌18h。高真空下蒸發溶劑并將反應混合物重溶于甲醇。通過制備型薄層色譜采用7.5:5:2.5:90的MeOH:Et3N:CH2Cl2混合物作為流動相進行純化。產量=35mg(58%).1H NMR(CD3OD,400MHz)δ8.57(s,1H),8.07(d,J=8.2Hz,1H),7.34(d,J=8.2Hz,1H),7.22(d,J=8.2Hz,2H),6.97(dd,J1=2.7Hz,J2=9.6Hz,2H),6.88(d,J=2.7Hz,2H),5.61-‐5.43(m,2H),3.94(d,J=4.1Hz,2H),3.69-‐3.55(m,6H),3.31(s,2H),2.41-‐2.31(m,2H),2.27-‐2.14(m,2H),2.03-‐1.95(m,1H),1.90(s,2H),1.84-1.71(m,4H),1.61-‐1.50(m,2H).13C NMR(CD3OD,100MHz)δ173.49,172.35,169.46,161.47,159.04,158.71,142.12,136.97,136.88,132.70,132.57,129.84,129.65,115.02,114.83,97.55,77.65,69.44,41.40,41.00,35.58,33.62,30.88,29.10.HRMS(ESI)m/z:計算C37H43N4O6[M+1]+639.3183;實測639.3151.
實施例15羅丹明釋放產物
將羅丹明-NHS酯(20.0mg,0.0379mmol)和N-Boc-乙二胺(13.2mg,0.0758mmol)溶于CH2Cl2(5mL)。加入三乙胺(51L,0.379mmol)并在室溫攪拌18h。高真空下蒸發溶劑并將反應混合物重溶于甲醇。通過制備型薄層色譜采用10:5:85的MeOH:Et3N:CH2Cl2混合物作為流動相進行純化。分離產物用4:1的CH2Cl2:TFA(5mL)混合物處理1h。產量=18mg(81%)。1H NMR(CD3OD,400MHz).8.83(s,1H),8.32(d,J=9.6Hz,1H),7.55(d,J=8.1Hz,1H),7.13(d,J=9.6Hz,2H),7.05(dd,J1=6.8Hz,J2=2.7Hz,2H),6.96(d,J=2.7Hz,2H),3.77(t,J=5.4Hz,,2H),3.30(s,12H),3.22-3.17(m,2H)。13C NMR(CD3OD,100MHz).169.28,167.47,160.69,159.10,138.52,132.63,132.11,132.02,131.67,115.71,114.85,97.63,41.08,40.97,39.06。HRMS(ESI)m/z:計算C27H29N4O4[M+1]+473.2189;實測473.2141.
實施例16可釋放的TCO-萬古霉素
將萬古霉素(50.0mg,0.0345mmol)和三乙胺(20μL,0.145mmol)溶于水(1mL)中。加入(E)-環辛-2-烯基4-硝基苯基碳酸酯(19.0mg,0.0652mmol)的DMF(25μL)溶液。在40℃攪拌反應混合物18h。通過45μm PTFE膜濾掉沉淀。上清液經HPLC純化從而獲得標題化合物。梯度10-60%CH3CN的水溶液用于HPLC純化。產量=3mg(5.5%)。1H NMR(CD3OD,400MHz)δ7.61(s,1H),7.55-7.38(m,2H),7.19(bs,1H),7.03(s,1H),6.81(bs,1H),6.44(s,1H),6.38(s,1H),6.05(bs,1H),5.55-5.37(m,2H),5.32(s,1H),5.23(s,1H),4.68(s,36H),4.52-4.31(m,2H),4.15(s,1H),4.00(t,J=8.2Hz,1H),3.85-3.61(m,3H),3.56(t,J=8.2Hz,1H),3.35(s,1H),2.67(s,4H),2.03-1.88(m,2H),1.75-1.39(m,3H),1.33(s,3H),1.05(s,3H),0.75(dd,J1=8.2Hz,J2=13.7Hz,6H)。HRMS(ESI)m/z:計算C75H87Cl2N9O26[M+1]+1602.4660;實測1602.5256.
實施例17可釋放的TCO-達托霉素
達托霉素(100mg,0.062mmol)溶于水(1mL)中。加入(E)-環辛-2-烯基4-硝基苯基碳酸酯(36mg,0.123mmol)的DMF(50μL)溶液,隨后加入三乙胺(167mg,1.65mmol)。室溫攪拌反應混合物18小時。使用半制備型菲羅門(Phenomenex)Luna 5u C18(2)柱和梯度10-65%CH3CN的水溶液經HPLC純化后獲得標題產物,為白色泡沫。產量=44mg(40%)。1H NMR(CD3OD,400MHz)δ7.66(bs,2H),7.60-7.46(m,2H),7.39(s,1H),7.25-7.11(m,2H),7.02(s,1H),6.96(s,1H),6.83-6.78(m,1H),6.56(s,1H),6.00-5.68(m,4H),5.68-5.57(m,1H),5.54(s,1H),5.51-5.26(m,8H),5.14(bs,1H),4.96(bs,1H),4.67(s,1H),4.64(s,1H),4.58(s,1H),4.14(s,1H),3.88-3.79(m,2H),3.78-3.71(m,1H),3.59-3.43(m,2H),3.19(q,J=8.2Hz,7H),2.95(bs,4H),2.59-2.32(m,5H),2.23-1.76(m,15H),1.56-1.44(m,8H),1.30(t,J=8.1Hz,10H),1.23-1.16(m,5H),1.00-0.87(m,9H)。MS(ESI)m/z:計算C75H87Cl2N9O26[M+H+MeCN]+1813.82;實測1813.80.
實施例18可釋放的TCO-阿霉素
根據Versteegen,R.M.等.,Angew.Chem.Int.Ed.2013,52,14112-14116所描述合成(E)-環辛烯阿霉素偶聯物。1H NMR(CDCl3)譜圖與公開數據相符。
實施例19可釋放的TCO-環腺苷酸
環腺苷酸(80mg,0.243mmol)和(E)-環辛-2-烯基4-硝基苯基碳酸酯(142mg,0.486mmol)溶于無水DMF(8mL)。加入4-二甲基氨基吡啶(238mg,1.94mmol)并在30℃攪拌反應混合物18h。高真空下去除溶劑,采用15%MeOH的二氯甲烷溶液作為流動相經制備柱色譜后獲得標題產物,為白色泡沫。產量=45mg(38%)。1H NMR(CDCl3,400MHz)δ7.61(s,1H),7.57-7.41(m,2H),7.19(bs,1H),7.03(s,1H),6.81(bs,1H),6.44(s,1H),6.38(s,1H),6.06(bs,1H),5.53-5.39(m,2H),5.32(s,2H),5.24(s,1H),4.48(bs,2H),4.16(s,1H),4.00(t,J=6.8Hz,1H),3.83-3.61(m,3H),3.60-3.50(m,1H),3.35(s,1H),2.68(s,4H),2.03-1.90(m,2H),1.75-1.40(m,3H),1.33(s,3H),1.05(s,3H),0.74(dd,J1=8.2Hz,J2=13.6Hz,6H)。HRMS(ESI)m/z:計算C19H25N5O8P[M+1]+482.1441;實測482.1461.
實施例20可釋放的TCO-化合物3
化合物3(132mg,0.413mmol)和(E)-環辛-2-烯基4-硝基苯基碳酸酯(80mg,0.275mmol)溶于無水DMF(5mL)。加入三乙胺(167mg,1.65mmol)并在室溫攪拌反應混合物18小時。高真空下去除溶劑,采用10%MeOH的二氯甲烷溶液作為流動相經制備柱色譜后獲得標題產物,為白色泡沫。產量=70mg(36%)。1H NMR(CD3OD,400MHz)δ8.28(s,1H),5.93(d,J=6.9Hz,1H),5.89-5.78(m,1H),5.54(bs,1H),5.28(bs,1H),4.73(t,J=5.5Hz,1H),4.37-4.31(m,1H),4.23-4.13(m,3H),3.92(d,J=10.9Hz,1H),3.73(d,J=12.3Hz,1H),3.65-3.40(m,1H),3.35(s,1H),3.31(s,1H),2.47-2.30(m,1H),2.10-1.75(m,4H),1.75-1.57(m,2H),1.53(m,1H),1.26(bs,1H),1.18-1.06(m,2H),0.91-0.74(m,1H)。HRMS(ESI)m/z:計算C22H28N6O6[M+1]+473.2149;實測473.2117.
盡管為了清楚理解的目的以說明和舉例的方式相當詳細描述前述發明,但本領域的技術人員將理解,在所附權利要求的范圍內可以作出某些改變和修改。另外,本文提供的各參考通過引用以其全部內容并入本文,如同通過引用單獨并入各參考相同的程度。本申請和本文提供的參考之間存在沖突之處,以本申請為準。
- 還沒有人留言評論。精彩留言會獲得點贊!