印刷布線板的制造方法與流程
本發明涉及印刷布線板的制造方法、用于所述制造方法的粘接片、和疊層結構體、以及具備通過所述制造方法制造的印刷布線板的半導體裝置。
背景技術:
作為印刷布線板的制造方法,已知采用交替重疊絕緣層和導體層的堆疊(buildup)方式的制造方法。采用的堆疊方式的制造方法中,一般絕緣層通過使樹脂組合物熱固化而形成。多層印刷布線板中,設有通過這樣的堆疊方式形成的多個堆疊層,隨著布線的進一步微細化和高密度化,需要對于導體層呈現良好的剝離強度的絕緣層。
作為這樣的絕緣層,例如專利文獻1中揭示了印刷布線板的制造方法,該方法包括:使用包含加熱時顯示特定的膨脹特性的支撐體的粘接片,并在熱固化后除去支撐體的工序。
現有技術文獻
專利文獻
專利文獻1:日本特開2015-162635號公報。
技術實現要素:
發明所要解決的技術問題
本發明人發現,在將粘接片接合于內層基板并在熱固化后剝離支撐體的制造方法中,熱固化工序中支撐體膨脹、收縮,因而樹脂組合物層也膨脹、收縮,樹脂組合物層表面產生固化不均,或者樹脂組合物層的厚度產生偏差(波動,undulation),因此印刷布線板的成品率下降。
因此,本發明的課題在于提供即使在帶有支撐體的狀態下將樹脂組合物層熱固化而形成絕緣層,也抑制了固化不均和波動的產生的印刷布線板的制造方法、用于所述制造方法的粘接片、和疊層結構體、以及具備通過所述制造方法制造的印刷布線板的半導體裝置。
解決技術問題用的技術方案
本發明人發現通過下述構成可抑制固化不均和波動,從而完成了本發明。
即,本發明包括以下的內容,
[1]印刷布線板的制造方法,該方法包括:
(a)準備具備支撐體和設置于該支撐體上的樹脂組合物層的粘接片的工序、
(b)以樹脂組合物層與內層基板接合的方式將粘接片疊層于內層基板的工序、以及
(c)通過將粘接片從t1(℃)加熱至t2(℃)而進行熱固化,形成絕緣層的工序,
其中,將(eamd-ebmd)與(eatd-ebtd)之和((eamd-ebmd)+(eatd-ebtd))設為x、將120℃以上時的樹脂組合物層的最低熔融粘度設為y(泊)時,以滿足y>2700x的關系的方式進行熱固化,
所述(eamd-ebmd)是,從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebmd(%)之差,
所述(eatd-ebtd)是,從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebtd(%)之差;
[2]如[1]所述的印刷布線板的制造方法,其中,滿足y>2700x>300的關系;
[3]如[1]或[2]所述的印刷布線板的制造方法,其中,x為4以下;
[4]如[1]~[3]中的任一項所述的印刷布線板的制造方法,其中,y為4000泊以上;
[5]印刷布線板的制造方法,該方法包括:
(a)準備具備支撐體和設置于該支撐體上的樹脂組合物層的粘接片的工序、
(b)以樹脂組合物層與內層基板接合的方式將粘接片疊層于內層基板的工序、以及
(c)通過將粘接片從t1(℃)加熱至t2(℃)而進行熱固化,形成絕緣層的工序,
其中,以(eamd-ebmd)與(eatd-ebtd)之和((eamd-ebmd)+(eatd-ebtd))為4以下、t1(℃)以上時的樹脂組合物層的最低熔融粘度為4000泊以上的方式進行熱固化,
所述(eamd-ebmd)是,從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebmd(%)之差,
所述(eatd-ebtd)是,從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebtd(%)之差;
[6]如[1]~[5]中的任一項所述的印刷布線板的制造方法,其中,t1滿足50℃≤t1≤150℃的關系;
[7]如[1]~[6]中的任一項所述的印刷布線板的制造方法,其中,t1滿足120℃≤t1≤150℃的關系;
[8]如[1]~[7]中的任一項所述的印刷布線板的制造方法,其中,t2滿足150℃≤t2≤240℃的關系;
[9]如[1]~[8]中的任一項所述的印刷布線板的制造方法,其中,工序(c)中,將粘接片在t1(℃)加熱后,進而在t2(℃)加熱,進行熱固化;
[10]如[1]~[9]中的任一項所述的印刷布線板的制造方法,其中,支撐體由塑料膜形成;
[11]半導體裝置,其具備通過[1]~[10]中的任一項所述的印刷布線板的制造方法制造的印刷布線板;
[12]疊層結構體,其具有內層基板、設置于該內層基板的絕緣層、以及接合于該絕緣層的支撐體,其中,在從支撐體的端部相對于支撐體的尺寸除去5%后的范圍內,該絕緣層的厚度的偏差為1μm以下;
[13]粘接片,其具備支撐體、和設置于該支撐體上的樹脂組合物層,
其中,通過將粘接片從t1(℃)加熱至t2(℃)而進行熱固化時,
將(eamd-ebmd)與(eatd-ebtd)之和((eamd-ebmd)+(eatd-ebtd))設為x、將120℃以上時的樹脂組合物層的最低熔融粘度設為y(泊)時,滿足y>2700x的關系,
所述(eamd-ebmd)是,從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebmd(%)之差,
所述(eatd-ebtd)是,從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebtd(%)之差。
發明的效果
如果采用本發明,則可提供即使在帶有支撐體的狀態下將樹脂組合物層熱固化而形成絕緣層、也抑制了固化不均和波動的產生的印刷布線板的制造方法、用于所述制造方法的粘接片、和疊層結構體、以及具備通過所述制造方法制造的印刷布線板的半導體裝置。
附圖說明
圖1是示出加熱時的支撐體的td方向和md方向上的膨脹行為的示意圖;
圖2是示出加熱時的支撐體的td方向和md方向上的膨脹行為的示意圖;
圖3是疊層結構體的示意性俯視圖;
圖4是疊層結構體的切割端面的示意圖。
具體實施方式
<術語的說明>
本發明中,對于支撐體所說的“md方向(加工方向,machinedirection)”是指制造支撐體時的支撐體的長邊方向,即制造時的長條的支撐體的運送方向。此外,對于支撐體所說的“td方向(橫向,transversedirection)”是指制造支撐體時的支撐體的寬度方向,是與md方向垂直的方向。應予說明,md方向和td方向都是相對于支撐體的厚度方向垂直的方向。
本發明中,支撐體的md方向或td方向上的支撐體的“膨脹系數”是指,將支撐體在規定的加熱條件下進行加熱時的支撐體的md方向或td方向上的支撐體的長度(尺寸)的增加比例(%)。對于支撐體的膨脹系數(%)而言,在將初始長度(即,加熱開始的時刻的支撐體的長度)設為l0、加熱規定時間后的支撐體的長度設為l時,通過算式:(l-l0)/l0×100求得。膨脹系數為正值時表示支撐體因加熱而膨脹,膨脹系數為負值時表示支撐體因加熱而收縮。支撐體的膨脹系數(%)可通過使用熱機械分析裝置測定在規定的加熱條件下進行加熱時的支撐體的md方向或td方向上的支撐體的長度的變化來求得。作為熱機械分析裝置,可例舉例如理學(rigaku)株式會社制“thermoplustma8310”、seikoinstruments株式會社制“tma-ss6100”。
應予說明,該加熱條件為特定支撐體的膨脹系數等特性時的條件,與后述的“將樹脂組合物層熱固化而形成絕緣層的工序(c)”中的條件可以一致,也可以不一致。
[粘接片]
對本發明的印刷布線板的制造方法進行詳細說明之前,對本發明的印刷布線板的制造方法中所使用的“粘接片”進行說明。
本發明的第一實施方式的粘接片是具備支撐體以及設置于該支撐體上的樹脂組合物層的粘接片,其中,通過將粘接片從t1(℃)加熱至t2(℃)而使其熱固化時,將(eamd-ebmd)與(eatd-ebtd)之和((eamd-ebmd)+(eatd-ebtd))設為x、將120℃以上時的樹脂組合物層的最低熔融粘度設為y(泊)時,滿足y>2700x的關系,所述(eamd-ebmd)是從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebmd(%)之差,所述(eatd-ebtd)是從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebtd(%)之差。
本發明的第二實施方式的粘接片是具備支撐體以及設置于該支撐體上的樹脂組合物層的粘接片,其中,通過將粘接片從t1(℃)加熱至t2(℃)而使其熱固化時,(eamd-ebmd)與(eatd-ebtd)之和((eamd-ebmd)+(eatd-ebtd))為4以下、t1(℃)以上時的樹脂組合物層的最低熔融粘度為4000泊以上,所述(eamd-ebmd)是從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebmd(%)之差,所述(eatd-ebtd)是從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebtd(%)之差。
在此,t1(℃)<t2(℃)。
一般來說,支撐體被加熱后會膨脹或收縮。加熱時的膨脹和/或收縮的程度根據支撐體的種類而不同,由于其制造工序(例如,支撐體的構成材料的選擇、運送的支撐體的卷取時(運送時)施加的張力等),支撐體存在被加熱后在md方向比td方向更容易收縮,在td方向比md方向更容易膨脹的傾向。
本發明中,在通過調整熱固化時的加熱條件來控制支撐體的膨脹、收縮的同時,從抑制伴隨支撐體的膨脹、收縮的固化不均和波動的形成的觀點來看,還控制樹脂組合物層的最低熔融粘度。
以下,對第一和第二實施方式的粘接片具備的支撐體及樹脂組合物層進行說明。
<支撐體>
參照圖1和圖2對本發明的實施方式所涉及的支撐體的膨脹行為的大致情況進行說明。
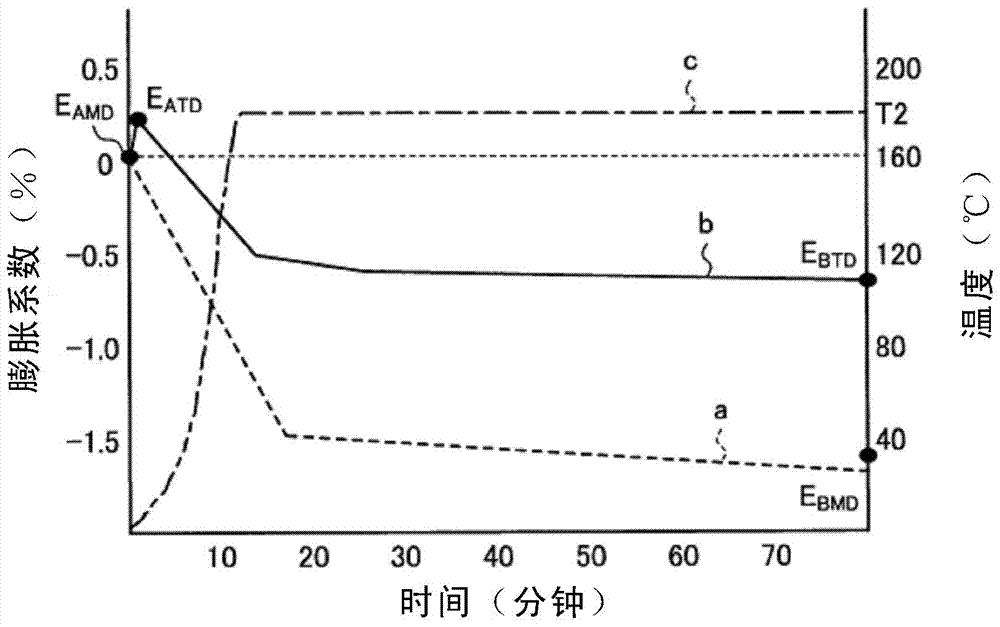
圖1和圖2是示意性地示出進行熱固化而形成絕緣層時的支撐體的td方向和md方向上的膨脹行為的圖。圖1和圖2中,左側縱軸表示支撐體的td方向和md方向上各自的膨脹系數(%),右側縱軸表示加熱溫度(℃),橫軸表示加熱時間(分鐘)。此外,曲線a表示md方向的膨脹行為,曲線b表示td方向的膨脹行為,曲線c表示經時的溫度曲線。
圖1是表示通過從室溫(例如20℃)升溫至t1(℃)、保持t1(℃)30分鐘后、從t1(℃)升溫至t2(℃)、在t2(℃)加熱30分鐘的所謂2步(2階段)加熱來進行熱固化時的支撐體的膨脹行為的圖。
對于md方向的支撐體的膨脹行為,由圖1可知,從室溫升溫至t1(℃)的過程(加熱時間0分鐘~10分鐘的區間)中,膨脹系數下降而成為負值,保持t1(℃)30分鐘的過程(加熱時間10分鐘~40分鐘的區間)中,膨脹系數也平穩地下降。接著,從t1(℃)升溫至t2(℃)的過程(加熱時間40分鐘~50分鐘的區間)中,膨脹系數大幅下降,保持t2(℃)30分鐘的過程(加熱時間50分鐘~80分鐘的區間)中,膨脹系數平穩地下降。
從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)是指從t1(℃)升溫至t2(℃)的過程中的md方向的支撐體的最大膨脹系數eamd(%)。圖1的形態中,最大膨脹系數eamd(%)為開始從t1(℃)至t2(℃)的加熱的時刻(加熱時間40分鐘的時刻)的md方向的支撐體的膨脹系數。
加熱結束的時刻的t2(℃)時的md方向的支撐體的膨脹系數ebmd(%)是指加熱結束的時刻的溫度t2(℃)時的md方向的支撐體的膨脹系數。圖1的形態中,膨脹系數ebmd(%)為t2(℃)的加熱結束的時刻(加熱時間80分鐘的時刻)的md方向的支撐體的膨脹系數。
對于td方向的支撐體的膨脹行為,由圖1可知,從室溫升溫至t1(℃)的過程(加熱時間0分鐘~10分鐘的區間)中,膨脹系數一度稍稍升高,但達到t1(℃)的時刻膨脹系數成為負值,在t1(℃)加熱30分鐘的過程(加熱時間10分鐘~40分鐘的區間)中,膨脹系數平穩地下降。接著,從t1(℃)升溫至t2(℃)的過程(加熱時間40分鐘~50分鐘的區間)中,膨脹系數大幅下降,在t2(℃)加熱30分鐘的過程(加熱時間50分鐘~80分鐘的區間)中,膨脹系數平穩地下降。
從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)是指從t1(℃)升溫至t2(℃)的過程中的td方向的支撐體的最大膨脹系數。圖1的形態中,最大膨脹系數eatd(%)為開始從t1(℃)至t2(℃)的加熱的時刻(加熱時間40分鐘的時刻)的支撐體的膨脹系數。
加熱結束的時刻的t2(℃)時的td方向的支撐體的膨脹系數ebtd(%)是指加熱結束的時刻的溫度t2(℃)時的td方向的支撐體的膨脹系數。圖1的形態中,膨脹系數ebtd(%)為t2(℃)的加熱結束的時刻(加熱時間80分鐘的時刻)的td方向的支撐體的膨脹系數。
由圖1的曲線a和曲線b可知,膨脹系數ebtd不會超過最大膨脹系數eatd,膨脹系數ebmd也不會超過最大膨脹系數eamd。
作為從t1(℃)升溫至t2(℃)的過程中的升溫速度,較好是1.5℃/分鐘~30℃/分鐘,更好是2℃/分鐘~30℃/分鐘,進一步更好是4℃/分鐘~20℃/分鐘,再進一步更好是4℃/分鐘~10℃/分鐘。從室溫升溫至t1(℃)的過程中的升溫速度也同樣。
圖2是表示通過從室溫(例如20℃)升溫至t2(℃)、在t2(℃)加熱65分鐘的所謂1步(1階段)加熱來進行熱固化時的支撐體的膨脹行為的圖。
對于md方向的支撐體的膨脹行為,由圖2可知,從室溫升溫至t2(℃)的過程(加熱時間0分鐘~15分鐘的區間)中,膨脹系數大幅下降而成為負值,達到t2(℃)后(加熱時間15分鐘~20分鐘的區間)膨脹系數也下降。保持t2(℃)65分鐘的過程(加熱時間15分鐘~80分鐘的區間)中,膨脹系數平穩地下降。
在1步加熱中,t1(℃)可以是室溫。因此,通過1步加熱使粘接片熱固化的情況下,從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)是指從室溫升溫至t2(℃)的過程中的md方向的支撐體的最大膨脹系數。圖2的形態中,最大膨脹系數eamd(%)為加熱時間0分鐘的時刻的md方向的支撐體的膨脹系數。
通過1步加熱使粘接片熱固化的情況下,加熱結束的時刻的t2(℃)時的md方向的支撐體的膨脹系數ebmd(%)也是指t2(℃)的加熱結束的時刻的md方向的支撐體的膨脹系數。圖2的形態中,膨脹系數ebmd(%)為t2(℃)的加熱結束的時刻(加熱時間80分鐘的時刻)的md方向的支撐體的膨脹系數。
對于td方向的支撐體的膨脹行為,由圖2可知,從室溫升溫至t2(℃)的過程(加熱時間0分鐘~15分鐘的區間)中,膨脹系數一度稍稍升高,但隨著接近t2(℃),膨脹系數大幅下降,保持t2(℃)65分鐘的過程(加熱時間15分鐘~80分鐘的區間)中,膨脹系數平穩地下降。
通過1步加熱使粘接片熱固化的情況下,從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)是指從室溫升溫至t2(℃)的過程中的td方向的支撐體的最大膨脹系數。圖2的形態中,最大膨脹系數eatd(%)為加熱時間3分鐘的時刻的td方向的支撐體的膨脹系數。
通過1步加熱使粘接片熱固化的情況下,加熱結束的時刻的t2(℃)時的td方向的支撐體的膨脹系數ebtd(%)也是指t2(℃)的加熱結束的時刻的td方向的支撐體的膨脹系數。圖2的形態中,膨脹系數ebtd(%)為t2(℃)的加熱結束的時刻(加熱時間80分鐘的時刻)的td方向的支撐體的膨脹系數。
由圖2的曲線a和曲線b可知,膨脹系數ebtd不會超過最大膨脹系數eatd,膨脹系數ebmd也不會超過最大膨脹系數eamd。
作為從室溫升溫至t2的過程中的升溫速度,較好是1.5℃/分鐘~30℃/分鐘,更好是2℃/分鐘~30℃/分鐘,進一步更好是4℃/分鐘~20℃/分鐘,再進一步更好是4℃/分鐘~10℃/分鐘。
作為第一和第二實施方式中的最大膨脹系數eamd(%)與膨脹系數ebmd(%)的差(eamd-ebmd),可設為0.05以上、0.1以上或0.3以上等。對于上限,較好是3以下,更好是2.5以下,進一步更好是2以下。
作為第一和第二實施方式中的最大膨脹系數eatd(%)與膨脹系數ebtd(%)的差(eatd-ebtd),可設為0.05以上、0.1以上或0.2以上等。對于上限,較好是3以下,更好是2.5以下,進一步更好是2以下。
第一實施方式的粘接片中的x((eamd-ebmd)+(eatd-ebtd))無特別限定,較好是4以下,更好是3.9以下,進一步更好是3.8以下或3.7以下。下限可設為0.01以上、0.5以上或0.8以上等。此外,2700x較好是10800以下,更好是10530以下,進一步更好是10260以下或9990以下。下限較好是27以上,更好是1350以上,進一步更好是2160以上。
從有效地抑制波動的觀點來看,第一實施方式的粘接片中的關系式(y>2700x)中的2700x較好是超過300。即,較好是滿足y>2700x>300的關系。
第二實施方式的粘接片中的(eamd-ebmd)+(eatd-ebtd)為4以下,較好是3.9以下,更好是3.8以下,進一步更好是3.7以下。對于下限,可設為0.01以上、0.5以上或0.8以上等。
本發明中,支撐體的低于t1(℃)時的膨脹系數無特別限定,將不損害本發明的目的作為條件,可具有超過0%的一定程度的膨脹系數。這是因為,即使假設在低于t1(℃)的溫度下支撐體膨脹而形成固化不均和波動,之后支撐體的膨脹系數變為0%以下的情況下,也可將所形成的固化不均和波動通過后述的樹脂組合物層的熔融而以自調整的方式平坦化。但是,為了獲得該自調整的平坦化的效果,低于t1(℃)時的膨脹系數較好是2%以下。
通常,支撐體開始急劇膨脹的溫度為120℃左右。支撐體的md方向的膨脹系數達到最大時的溫度無特別限定,為60℃以上、80℃以上。支撐體的td方向的膨脹系數達到最大時的溫度無特別限定,為60℃以上、80℃以上。
圖1的形態、即進行采用2步的加熱時,作為t1(℃),只要低于t2(℃)即可,無特別限定,從減少因溶劑成分的發泡導致的空隙的產生的觀點來看,較好是滿足50℃≤t1≤150℃的關系,更好是滿足80℃≤t1≤150℃的關系,進一步更好是滿足120℃≤t1≤150℃的關系。
圖2的形態、即進行采用1步的加熱時,作為t1(℃),只要低于t2(℃)即可,無特別限定,較好是滿足0℃≤t1≤50℃的關系,更好是滿足5℃≤t1≤40℃的關系,進一步更好是滿足10℃≤t1≤30℃的關系。
作為t2(℃),只要溫度比t1(℃)高,樹脂組合物層發生熱固化即可,無特別限定,較好是滿足150℃≤t2≤240℃的關系,更好是滿足155℃≤t2≤200℃的關系,進一步更好是滿足170℃≤t2≤180℃的關系。
作為支撐體,從為輕量的同時在制造印刷布線板時顯示所需的強度的觀點來看,可優選使用由塑料材料形成的膜(以下也簡稱“塑料膜”)。作為塑料材料,可例舉例如聚對苯二甲酸乙二醇酯(稱為“pet”)、聚萘二甲酸乙二醇酯(稱為“pen”)等聚酯、聚碳酸酯(稱為“pc”)、聚甲基丙烯酸甲酯(pmma)等丙烯酸類、環狀聚烯烴、三乙酰纖維素(tac)、聚醚硫化物(pes)、聚醚酮、聚酰亞胺等。作為支撐體,其中較好是聚對苯二甲酸乙二醇酯、聚萘二甲酸乙二醇酯,特別好是價格便宜的聚對苯二甲酸乙二醇酯。
本發明的一種優選實施方式中,對由塑料膜等形成的支撐體進行預加熱處理,制備控制了膨脹和收縮的支撐體。預加熱處理可根據塑料材料的種類、制造時施加的張力(拉伸)的有無、拉伸的軸方向、拉伸的程度、拉伸后的熱處理條件等調整條件進行,結果使得差(eamd-ebmd)與差(eatd-ebtd)之和x滿足所需的范圍。
塑料膜采用長條的塑料膜的情況下,作為用于控制支撐體的膨脹和收縮的預加熱處理,可例舉例如在塑料膜的md方向和td方向中的一方或兩方施加張力并進行加熱的處理。
使用長條的塑料膜的情況下,通常,通過制造時的使用運送輥等輥的運送而在md方向施加規定的張力,因此有時通過僅在td方向施加張力并進行加熱,就可獲得差(eamd-ebmd)及差(eatd-ebtd)滿足所需的值的支撐體。
如果是md方向,可通過調整鋪開在多根輥間的塑料膜上施加的張力來施加規定的張力。此外,在td方向施加張力時,可通過目前公知的任意的合適手段來實施。此外,在td方向施加張力時,可使用具有目前公知的結構的拉幅機等來施加規定的張力。
此外,例如可利用重物的重量和重力在塑料膜的md方向或td方向施加規定的張力。具體來說,可按照td方向和md方向中要調整的方向與垂直方向一致的方式將塑料膜的要調整的方向的一側的端緣部通過任意合適的粘接構件(例如聚酰亞胺(kapton)膠帶、ptfe膠帶、玻璃布膠帶)固定于例如支承棒等,以張力均勻地施加到整個塑料膜的方式吊起。然后,在對置的要調整的方向的另一側的端緣部以張力均勻地施加到整個塑料膜的方式通過任意合適的粘接構件連接金屬板等重物而通過重物的重量來施加張力,同時進行加熱,從而可進行預加熱處理。
對于塑料膜上施加的張力的大小,可考慮塑料膜的材料、膨脹系數、樹脂組合物的組成等而設定為任意合適的張力。例如,可例舉將張力設為1gf/cm2~40gf/cm2的條件。
一個實施方式中,將塑料膜的玻璃化轉變溫度設為tg時,預加熱處理的加熱溫度較好是(tg+50)℃以上,更好是(tg+60)℃以上,進一步更好是(tg+70)℃以上,再進一步更好是(tg+80)℃以上或(tg+90)℃以上。只要低于塑料膜的熔點,加熱溫度的上限較好是(tg+115)℃以下,更好是(tg+110)℃以下,進一步更好是(tg+105)℃以下。
支撐體例如為pet膜的情況下,預加熱處理的加熱溫度較好是100℃以上,更好是110℃以上,進一步更好是120℃以上,再進一步更好是125℃以上或130℃以上。加熱溫度的上限較好是195℃以下,更好是190℃以下,進一步更好是185℃以下、180℃以下或175℃以下。
從控制支撐體的膨脹和收縮的觀點來看,加熱時間根據加熱溫度適當決定即可。一個實施方式中,加熱時間較好是1分鐘以上,更好是2分鐘以上,進一步更好是5分鐘以上、10分鐘以上或15分鐘以上。加熱時間的上限也取決于加熱溫度,但較好是120分鐘以下,更好是90分鐘以下,進一步更好是60分鐘以下。
實施預加熱處理時的氣氛無特別限定,可例舉例如大氣氣氛、惰性氣體氣氛(氮氣氣氛、氦氣氣氛、氬氣氣氛等),從可簡便地制備支撐體的觀點來看,較好是大氣氣氛。
預加熱處理可在減壓下、常壓下、加壓下中的任一條件下實施,從可簡便地制備支撐體的觀點來看,較好是在常壓下實施。
支撐體可在與后述的樹脂組合物層接合的面實施消光處理、電暈處理。
此外,作為支撐體,可使用在與樹脂組合物層接合的面具有脫模層的帶脫模層的支撐體。作為用于帶脫模層的支撐體的脫模層的脫模劑,可例舉例如選自醇酸樹脂、聚烯烴樹脂、聚氨酯樹脂和有機硅樹脂的1種以上的脫模劑。帶脫模層的支撐體可使用市售品,可例舉例如作為具有以醇酸樹脂類脫模劑為主要成分的脫模層的pet膜的琳得科株式會社制的“sk-1”、“al-5”、“al-7”、東麗株式會社制的“lumirrort6am”等。
支撐體的厚度無特別限定,較好是在5μm~75μm的范圍內,更好是在10μm~60μm的范圍內,進一步更好是在10μm~45μm的范圍內。應予說明,使用帶脫模層的支撐體的情況下,較好是帶脫模層的支撐體整體的厚度在上述范圍內。
<樹脂組合物層>
粘接片具備的樹脂組合物層中所用的樹脂組合物無特別限定,只要其固化物具有足夠的硬度和絕緣性即可。作為樹脂組合物,可例舉例如包含固化性樹脂及其固化劑的組合物。作為固化性樹脂,可使用形成印刷布線板的絕緣層時所使用的目前公知的固化性樹脂,其中較好是環氧樹脂。因此,一個實施方式中,樹脂組合物包含(a)環氧樹脂、(b)固化劑及(c)無機填充材料。樹脂組合物可根據需要進一步包含熱塑性樹脂、固化促進劑、阻燃劑及有機填充材料。
以下,對可作為樹脂組合物的材料使用的各成分進行詳細說明。
-(a)環氧樹脂-
作為環氧樹脂,可例舉例如雙二甲酚(bixylenol)型環氧樹脂、雙酚a型環氧樹脂、雙酚f型環氧樹脂、雙酚s型環氧樹脂、雙酚af型環氧樹脂、二環戊二烯型環氧樹脂、三酚型環氧樹脂、萘酚酚醛型環氧樹脂(naphtholnovolacepoxyresin)、苯酚酚醛型環氧樹脂(phenolnovolacepoxyresin)、叔丁基兒茶酚型環氧樹脂、萘型環氧樹脂、萘酚型環氧樹脂、蒽型環氧樹脂、縮水甘油胺型環氧樹脂、縮水甘油酯型環氧樹脂、甲酚酚醛型環氧樹脂(cresolnovolacepoxyresin)、聯苯型環氧樹脂、線性脂肪族環氧樹脂、具有丁二烯結構的環氧樹脂、脂環族環氧樹脂、雜環型環氧樹脂、含螺環的環氧樹脂、環己烷二甲醇型環氧樹脂、亞萘基醚型環氧樹脂、三羥甲基型環氧樹脂、四苯基乙烷型環氧樹脂等。環氧樹脂可單獨使用1種,也可2種以上組合使用。從使平均線熱膨脹系數降低的觀點來看,(a)成分較好是含芳香族骨架的環氧樹脂,更好是選自雙酚a型環氧樹脂、雙酚f型環氧樹脂、萘型環氧樹脂、聯苯型環氧樹脂及二環戊二烯型環氧樹脂的1種以上,進一步更好是雙酚a型環氧樹脂、萘型環氧樹脂。芳香族骨架是指一般定義為芳香族的化學結構,也包括多環芳香族及芳香族雜環。
環氧樹脂較好是包括1分子中具有2個以上的環氧基的環氧樹脂。將環氧樹脂的不揮發成分設為100質量%的情況下,較好是至少50質量%以上為1分子中具有2個以上的環氧基的環氧樹脂。其中,較好是包括1分子中具有2個以上的環氧基且溫度20℃時呈液態的環氧樹脂(以下稱為“液態環氧樹脂”)和1分子中具有3個以上的環氧基且溫度20℃時呈固態的環氧樹脂(以下稱為“固態環氧樹脂”)。作為環氧樹脂,通過并用固態環氧樹脂和液態環氧樹脂,可獲得具有良好的撓性的樹脂組合物。此外,樹脂組合物的固化物的斷裂強度也提高。
作為液態環氧樹脂,較好是雙酚a型環氧樹脂、雙酚f型環氧樹脂、雙酚af型環氧樹脂、萘型環氧樹脂、縮水甘油酯型環氧樹脂、縮水甘油胺型環氧樹脂、苯酚酚醛型環氧樹脂、具有酯骨架的脂環族環氧樹脂、環己烷二甲醇型環氧樹脂、縮水甘油胺型環氧樹脂、及具有丁二烯結構的環氧樹脂,更好是縮水甘油胺型環氧樹脂、雙酚a型環氧樹脂、雙酚f型環氧樹脂、雙酚af型環氧樹脂及萘型環氧樹脂。作為液態環氧樹脂的具體例子,可例舉dic株式會社制的“hp4032”、“hp4032d”、“hp4032ss”(萘型環氧樹脂),三菱化學株式會社制的“828us”、“jer828el”、“825”、“epikote828el”(雙酚a型環氧樹脂)、“jer807”、“1750”(雙酚f型環氧樹脂)、“jer152”(苯酚酚醛型環氧樹脂)、“630”、“630lsd”(縮水甘油胺型環氧樹脂),新日鐵住金化學株式會社制的“zx1059”(雙酚a型環氧樹脂與雙酚f型環氧樹脂的混合品),nagasechemtex株式會社制的“ex-721”(縮水甘油酯型環氧樹脂),株式會社大賽璐制的“celloxide2021p”(具有酯骨架的脂環族環氧樹脂)、“pb-3600”(具有丁二烯結構的環氧樹脂),新日鐵化學株式會社制的“zx1658”、“zx1658gs”(液態1,4-縮水甘油基環己烷),三菱化學株式會社制的“630lsd”(縮水甘油胺型環氧樹脂)等。它們可單獨使用1種,也可2種以上組合使用。
作為固態環氧樹脂,較好是萘型四官能環氧樹脂、甲酚酚醛型環氧樹脂、二環戊二烯型環氧樹脂、三酚型環氧樹脂、萘酚型環氧樹脂、聯苯型環氧樹脂、亞萘基醚型環氧樹脂、蒽型環氧樹脂、雙酚a型環氧樹脂、四苯基乙烷型環氧樹脂,更好是萘型四官能環氧樹脂、萘酚型環氧樹脂、及聯苯型環氧樹脂。作為固態環氧樹脂的具體例子,可例舉dic株式會社制的“hp4032h”(萘型環氧樹脂)、“hp-4700”、“hp-4710”(萘型四官能環氧樹脂)、“n-690”(甲酚酚醛型環氧樹脂)、“n-695”(甲酚酚醛型環氧樹脂)、“hp-7200”(二環戊二烯型環氧樹脂)、“hp-7200hh”、“hp-7200h”、“exa-7311”、“exa-7311-g3”、“exa-7311-g4”、“exa-7311-g4s”、“hp6000”(亞萘基醚型環氧樹脂),日本化藥株式會社制的“eppn-502h”(三酚型環氧樹脂)、“nc7000l”(萘酚酚醛型環氧樹脂)、“nc3000h”、“nc3000”、“nc3000l”、“nc3100”(聯苯型環氧樹脂),新日鐵住金化學株式會社制的“esn475v”(萘型環氧樹脂)、“esn485”(萘酚酚醛型環氧樹脂),三菱化學株式會社制的“yx4000h”、“yx4000”、“yl6121”(聯苯型環氧樹脂)、“yx4000hk”(雙二甲酚型環氧樹脂)、“yx8800”(蒽型環氧樹脂),大阪燃氣化學株式會社制的“pg-100”、“cg-500”、三菱化學株式會社制的“yl7760”(雙酚af型環氧樹脂)、“yl7800”(芴型環氧樹脂)、三菱化學株式會社制的“jer1010”(固態雙酚a型環氧樹脂)、“jer1031s”(四苯基乙烷型環氧樹脂)等。它們可單獨使用1種,也可2種以上組合使用。
作為環氧樹脂,并用液態環氧樹脂和固態環氧樹脂的情況下,它們的量比(液態環氧樹脂:固態環氧樹脂)以質量比計較好是在1:0.1~1:15的范圍內。通過使液態環氧樹脂與固態環氧樹脂的量比在這樣的范圍內,可獲得i)以粘接片的形態使用時帶來適度的粘附性、ii)以粘接片的形態使用時能獲得足夠的撓性而操作性提高、以及iii)能得到具有足夠的斷裂強度的固化物等效果。從上述i)~iii)的效果的觀點來看,液態環氧樹脂與固態環氧樹脂的量比(液態環氧樹脂:固態環氧樹脂)以質量比計更好是在1:0.5~1:10的范圍內,進一步更好是在1:1~1:8的范圍內。
從獲得顯示良好的機械強度、絕緣可靠性的絕緣層的觀點來看,樹脂組合物中的環氧樹脂的含量較好是1質量%以上,更好是2質量%以上,進一步更好是3質量%以上。只要發揮本發明的效果,環氧樹脂的含量的上限無特別限定,但較好是60質量%以下,更好是50質量%以下,進一步更好是40質量%以下。
應予說明,本發明中,只要沒有另行明示,樹脂組合物中的各成分的含量為將樹脂組合物中的不揮發成分設為100質量%時的值。
環氧樹脂的環氧當量較好是50~5000,更好是50~3000,進一步更好是80~2000,再進一步更好是110~1000。通過使其在該范圍內,固化物的交聯密度充分,可帶來表面粗糙度小的絕緣層。應予說明,環氧當量可按照jisk7236進行測定,是包含1當量的環氧基的樹脂的質量。
環氧樹脂的重均分子量較好是100~5000,更好是250~3000,進一步更好是400~1500。在此,環氧樹脂的重均分子量為通過凝膠滲透色譜法(gpc)測定的聚苯乙烯換算的重均分子量。
-(b)固化劑-
作為固化劑,只有具有使環氧樹脂固化的功能即可,無特別限定,可例舉例如苯酚類固化劑、萘酚類固化劑、活性酯類固化劑、苯并噁嗪類固化劑、氰酸酯類固化劑及碳二亞胺類固化劑等。固化劑可單獨使用1種,也可2種以上并用。(b)成分較好是選自苯酚類固化劑、萘酚類固化劑、活性酯類固化劑、碳二亞胺類固化劑及氰酸酯類固化劑的1種以上。
作為苯酚類固化劑及萘酚類固化劑,從耐熱性和耐水性的觀點來看,較好是具有酚醛結構(novolacstructure)的苯酚類固化劑或者具有酚醛結構的萘酚類固化劑。此外,從與導體層的密合性的觀點來看,較好是含氮苯酚類固化劑,更好是含三嗪骨架的苯酚類固化劑。其中,從使耐熱性、耐水性及與導體層的密合性高度滿足的觀點來看,較好是含三嗪骨架的苯酚酚醛類固化劑。
作為苯酚類固化劑及萘酚類固化劑的具體例子,可例舉例如明和化成株式會社制的“meh-7700”、“meh-7810”、“meh-7851”、“meh-7851h”,日本化藥株式會社制的“nhn”、“cbn”、“gph”,新日鐵住金株式會社制的“sn170”、“sn180”、“sn190”、“sn475”、“sn485”、“sn495”、“sn375”、“sn395”,dic株式會社制的“td-2090”、“la-7052”、“la-7054”、“la-1356”、“la-3018-50p”、“la-3018”、“exb-9500”等。
從獲得與導體層的密合性良好的絕緣層的觀點來看,較好是活性酯類固化劑。作為活性酯類固化劑,無特別限定,一般優選使用苯酚酯類、苯硫酚酯類、n-羥基胺酯類、雜環羥基化合物的酯類等1分子中具有2個以上的反應活性高的酯基的化合物。該活性酯類固化劑較好是通過羧酸化合物和/或硫代羧酸化合物與羥基化合物和/或硫醇化合物的縮合反應獲得的活性酯類固化劑。特別是從耐熱性提高的觀點來看,較好是由羧酸化合物與羥基化合物獲得的活性酯類固化劑,更好是由羧酸化合物與苯酚化合物和/或萘酚化合物獲得的活性酯類固化劑。作為羧酸化合物,可例舉例如苯甲酸、乙酸、琥珀酸、馬來酸、衣康酸、鄰苯二甲酸、間苯二甲酸、對苯二甲酸、均苯四酸等。作為苯酚化合物或萘酚化合物,可例舉例如氫醌、間苯二酚、雙酚a、雙酚f、雙酚s、酚酞啉、甲基化雙酚a、甲基化雙酚f、甲基化雙酚s、苯酚、鄰甲酚、間甲酚、對甲酚、兒茶酚、α-萘酚、β-萘酚、1,5-二羥基萘、1,6-二羥基萘、2,6-二羥基萘、二羥基二苯酮、三羥基二苯酮、四羥基二苯酮、間苯三酚、苯三酚、二環戊二烯型二苯酚化合物、苯酚酚醛(phenolnovolac)等。在此,“二環戊二烯型二苯酚化合物”是指1分子二環戊二烯縮合2分子苯酚而得的二苯酚化合物。
具體來說,較好是含二環戊二烯型二苯酚結構的活性酯化合物、含萘結構的活性酯化合物、含苯酚酚醛的乙酰基化物的活性酯化合物、含苯酚酚醛的苯甲酰基化物的活性酯化合物,其中更好是含萘結構的活性酯化合物、含二環戊二烯型二苯酚結構的活性酯化合物。“二環戊二烯型二苯酚結構”表示由亞苯基-亞二環戊基-亞苯基形成的2價結構單元。
作為活性酯類固化劑的市售品,含二環戊二烯型二苯酚結構的活性酯化合物可例舉“exb9451”、“exb9460”、“exb9460s”、“hpc-8000-65t”、“hpc-8000h-65tm”、“exb-8000l-65tm”(dic株式會社制)等,含萘結構的活性酯化合物可例舉“exb9416-70bk”(dic株式會社制)等,含苯酚酚醛的乙酰基化物的活性酯化合物可例舉“dc808”(三菱化學株式會社制)等,含苯酚酚醛的苯甲酰基化物的活性酯化合物可例舉“ylh1026”(三菱化學株式會社制)等,作為苯酚酚醛的乙酰基化物的活性酯類固化劑可例舉“dc808”(三菱化學株式會社制)等,作為苯酚酚醛的苯甲酰基化物的活性酯類固化劑可例舉“ylh1026”(三菱化學株式會社制)、“ylh1030”(三菱化學株式會社制)、“ylh1048”(三菱化學株式會社制)等。
作為苯并噁嗪類固化劑的具體例子,可例舉昭和高分子株式會社制的“hfb2006m”、四國化成工業株式會社制的“p-d”、“f-a”。
作為氰酸酯類固化劑,可例舉例如雙酚a二氰酸酯、多酚氰酸酯、低聚(3-亞甲基-1,5-亞苯基氰酸酯)、4,4'-亞甲基雙(2,6-二甲基苯基氰酸酯)、4,4'-亞乙基二苯基二氰酸酯、六氟雙酚a二氰酸酯、2,2-雙(4-氰酸酯基)苯基丙烷、1,1-雙(4-氰酸酯基苯基甲烷)、雙(4-氰酸酯基-3,5-二甲基苯基)甲烷、1,3-雙(4-氰酸酯基苯基-1-(甲基亞乙基))苯、雙(4-氰酸酯基苯基)硫醚、和雙(4-氰酸酯基苯基)醚等二官能氰酸酯樹脂、由苯酚酚醛及甲酚酚醛等所衍生的多官能氰酸酯樹脂、這些氰酸酯樹脂部分三嗪化而得的預聚物等。作為氰酸酯類固化劑的具體例子,可例舉lonzajapan株式會社制的“pt30”和“pt60”(均為苯酚酚醛型多官能氰酸酯樹脂)、“ba230”、“ba230s75”(雙酚a二氰酸酯的一部分或全部被三嗪化而形成三聚體的預聚物)等。
作為碳二亞胺類固化劑的具體例子,可例舉日清紡化學(nisshinbochemical)株式會社制的“v-03”、“v-07”等。
環氧樹脂與固化劑的量比以[環氧樹脂的環氧基的總數]:[固化劑的反應基團的總數]的比例計較好是在1:0.01~1:2的范圍內,更好是在1:0.05~1:1.5的范圍內,進一步更好是在1:0.1~1:1的范圍內。在此,固化劑的反應基團是指活性羥基、活性酯基等,根據固化劑的種類而不同。此外,環氧樹脂的環氧基的總數是指對于所有的環氧樹脂將各環氧樹脂的固體成分質量除以環氧當量而得的值進行合計得到的值,固化劑的反應基團的總數是指對于所有的固化劑將各固化劑的固體成分質量除以反應基團當量而得的值進行合計得到的值。通過使環氧樹脂與固化劑的量比在這樣的范圍內,從而樹脂組合物的固化物的耐熱性進一步提高。
一個實施方式中,樹脂組合物包含前述的(a)環氧樹脂和(b)固化劑。樹脂組合物較好是分別作為(a)環氧樹脂包含液態環氧樹脂和固態環氧樹脂的混合物(液態環氧樹脂:固態環氧樹脂的質量比較好是1:0.1~1:15,更好是1:0.5~1:10,進一步更好是1:1.1~1:8),且作為(b)固化劑包含選自苯酚類固化劑、萘酚類固化劑、活性酯類固化劑、碳二亞胺類固化劑和氰酸酯類固化劑的1種以上。
樹脂組合物中的固化劑的含量無特別限定,較好是45質量%以下,更好是40質量%以下,進一步更好是35質量%以下。此外,下限無特別限定,較好是3質量%以上,更好是5質量%以上。
-(c)無機填充材料-
無機填充材料的材料無特別限定,可例舉例如二氧化硅、氧化鋁、玻璃、堇青石、硅氧化物、硫酸鋇、碳酸鋇、滑石、粘土、云母粉、氧化鋅、水滑石、勃姆石、氫氧化鋁、氫氧化鎂、碳酸鈣、碳酸鎂、氧化鎂、氮化硼、氮化鋁、氮化錳、硼酸鋁、碳酸鍶、鈦酸鍶、鈦酸鈣、鈦酸鎂、鈦酸鉍、氧化鈦、氧化鋯、鈦酸鋇、鈦酸鋯酸鋇、鋯酸鋇、鋯酸鈣、磷酸鋯和磷酸鎢酸鋯、碳化硅等。這些材料中,優選碳化硅、二氧化硅,特別優選二氧化硅。此外,作為二氧化硅,較好是球形二氧化硅。無機填充材料可單獨使用1種,也可2種以上組合使用。
無機填充材料的平均粒徑無特別限定,從使電路埋入性提高、獲得表面粗糙度小的絕緣層的觀點來看,較好是5μm以下,更好是2.5μm以下,進一步更好是2.2μm以下,再進一步更好是2μm以下。該平均粒徑的下限無特別限定,但較好是0.01μm以上,更好是0.05μm以上,進一步更好是0.1μm以上。作為具有這樣的平均粒徑的無機填充材料的市售品,可例舉例如株式會社admatechs制“yc100c”、“ya050c”、“ya050c-mje”、“ya010c”、電氣化學工業株式會社制“ufp-30”、株式會社德山制“シルフィルnss-3n”、“シルフィルnss-4n”、“シルフィルnss-5n”、株式會社admatechs制“sc2500sq”、“so-c6”、“so-c4”、“so-c2”、“so-c1”等。
無機填充材料的平均粒徑可通過基于米氏(mie)散射理論的激光衍射散射法進行測定。具體來說,可通過激光衍射散射式粒度分布測定裝置,以體積基準制成無機填充材料的粒度分布,將其中值粒徑作為平均粒徑來進行測定。測定樣品可優選使用通過超聲波使無機填充材料分散于甲基乙基酮中而得的樣品。作為激光衍射散射式粒度分布測定裝置,可使用株式會社島津制作所制“sald-2200”等。
從提高耐濕性和分散性的觀點來看,無機填充材料較好是通過選自氨基硅烷類偶聯劑、環氧基硅烷類偶聯劑、巰基硅烷類偶聯劑、烷氧基硅烷化合物、有機硅氮烷化合物、鈦酸酯類偶聯劑等的1種以上的表面處理劑進行了處理。作為表面處理劑的市售品,可例舉例如信越化學工業株式會社制“kbm403”(3-環氧丙氧基丙基三甲氧基硅烷)、信越化學工業株式會社制“kbm803”(3-巰基丙基三甲氧基硅烷)、信越化學工業株式會社制“kbe903”(3-氨基丙基三乙氧基硅烷)、信越化學工業株式會社制“kbm573”(n-苯基-3-氨基丙基三甲氧基硅烷)、信越化學工業株式會社制“sz-31”(六甲基二硅氮烷)、信越化學工業株式會社制“kbm-103”(苯基三甲氧基硅烷)、信越化學工業株式會社制“kbm-4803”(長鏈環氧型硅烷偶聯劑)等。表面處理劑可單獨使用1種,也可2種以上組合使用。
通過表面處理劑進行的表面處理的程度可通過無機填充材料的單位表面積的碳量進行評價。從無機填充材料的分散性提高的觀點來看,無機填充材料的單位表面積的碳量較好是0.02mg/m2以上,更好是0.1mg/m2以上,進一步更好是0.2mg/m2以上。另一方面,從樹脂清漆的熔融粘度和抑制片材形態下的熔融粘度的上升的觀點來看,較好是1mg/m2以下,更好是0.8mg/m2以下,進一步更好是0.5mg/m2以下。
無機填充材料的單位表面積的碳量可在將表面處理后的無機填充材料通過溶劑(例如甲基乙基酮(mek))進行清洗處理后進行測定。具體來說,向通過表面處理劑進行了表面處理的無機填充材料加入足量的mek作為溶劑,在25℃超聲波清洗5分鐘。除去上清液,使固體成分干燥后,可使用碳分析儀測定無機填充材料的單位表面積的碳量。作為碳分析儀,可使用株式會社堀場制作所制“emia-320v”等。
從獲得熱膨脹系數低的絕緣層的觀點來看,樹脂組合物中的無機填充材料的含量較好是30質量%以上,更好是35質量%以上,進一步更好是36質量%以上。從絕緣層的機械強度、特別是伸長率的觀點來看,上限較好是80質量%以下,更好是75質量%以下,進一步更好是70質量%以下。
-(d)熱塑性樹脂-
作為熱塑性樹脂,可例舉例如苯氧樹脂、聚乙烯醇縮醛樹脂、聚烯烴樹脂、聚丁二烯樹脂、聚酰亞胺樹脂、聚酰胺酰亞胺樹脂、聚醚酰亞胺樹脂、聚砜樹脂、聚醚砜樹脂、聚苯醚樹脂、聚碳酸酯樹脂、聚醚醚酮樹脂、聚酯樹脂,較好是苯氧樹脂。熱塑性樹脂可單獨使用1種,也可2種以上組合使用。
熱塑性樹脂的聚苯乙烯換算的重均分子量較好是8000~70000的范圍內,更好是10000~60000的范圍內,進一步更好是20000~60000的范圍內。熱塑性樹脂的聚苯乙烯換算的重均分子量通過凝膠滲透色譜法(gpc)測定。具體來說,熱塑性樹脂的聚苯乙烯換算的重均分子量可使用株式會社島津制作所制lc-9a/rid-6a作為測定裝置,使用昭和電工株式會社制shodexk-800p/k-804l/k-804l作為柱,使用氯仿等作為流動相,在40℃的柱溫下進行測定,使用標準聚苯乙烯的標準曲線算出。
作為苯氧樹脂,可例舉例如具有選自雙酚a骨架、雙酚f骨架、雙酚s骨架、雙酚乙酰苯骨架、酚醛骨架、聯苯骨架、芴骨架、二環戊二烯骨架、降冰片烯骨架、萘骨架、蒽骨架、金剛烷骨架、萜烯骨架和三甲基環己烷骨架的1種以上的骨架的苯氧樹脂。苯氧樹脂的末端可以是酚式羥基、環氧基等中的任一種官能團。苯氧樹脂可單獨使用1種,也可2種以上組合使用。作為苯氧樹脂的具體例子,可例舉三菱化學株式會社制的“1256”和“4250”(均為含雙酚a骨架的苯氧樹脂)、“yx8100”(含雙酚s骨架的苯氧樹脂)和“yx6954”(含雙酚乙酰苯骨架的苯氧樹脂),其它還可列舉新日鐵住金化學株式會社制的“fx280”和“fx293”、三菱化學株式會社制的“yx6954bh30”、“yx7553”、“yx7553bh30”、“yl7553bh30”、“yl7769bh30”、“yl6794”、“yl7213”、“yl7290”、“yl7891bh30”和“yl7482”等。
作為聚乙烯醇縮醛樹脂,可例舉例如聚乙烯醇縮甲醛樹脂、聚乙烯醇縮丁醛樹脂,較好是聚乙烯醇縮丁醛樹脂。作為聚乙烯醇縮醛樹脂的具體例子,可例舉例如denka株式會社制的“電化縮丁醛(denkabutyral)4000-2”、“電化縮丁醛5000-a”、“電化縮丁醛6000-c”、“電化縮丁醛6000-ep”,積水化學工業株式會社制的s-lecbh系列、bx系列(例如bx-5z)、ks系列(例如ks-1)、bl系列、bm系列等。
聚酰亞胺樹脂的具體例子,可例舉新日本理化株式會社制的“rikacoatsn20”和“rikacoatpn20”。作為聚酰亞胺樹脂的具體例子,還可例舉使二官能性羥基末端聚丁二烯、二異氰酸酯化合物和四元酸酐反應而得的線性聚酰亞胺(日本特開2006-37083號公報記載的聚酰亞胺)、含聚硅氧烷骨架的聚酰亞胺(日本特開2002-12667號公報和日本特開2000-319386號公報等中記載的聚酰亞胺)等改性聚酰亞胺。
作為聚酰胺酰亞胺樹脂的具體例子,還可例舉東洋紡績株式會社制的“vylomaxhr11nn”和“vylomaxhr16nn”。作為聚酰胺酰亞胺樹脂的具體例子,還可例舉日立化成工業株式會社制的“ks9100”、“ks9300”(含聚硅氧烷骨架的聚酰胺酰亞胺)等改性聚酰胺酰亞胺。
作為聚醚砜樹脂的具體例子,可例舉住友化學株式會社制的“pes5003p”等。
作為聚砜樹脂的具體例子,可例舉solvayadvancedpolymers株式會社制的聚砜“p1700”、“p3500”等。
作為聚苯醚樹脂的具體例子,可例舉三菱瓦斯化學株式會社制的低聚苯醚-苯乙烯樹脂“ope-2st1200”等。
其中,作為熱塑性樹脂,較好是苯氧樹脂、聚乙烯醇縮醛樹脂。因此,優選的一個實施方式中,熱塑性樹脂包括選自苯氧樹脂和聚乙烯醇縮醛樹脂的1種以上。
樹脂組合物包含熱塑性樹脂的情況下,熱塑性樹脂的含量較好是0.1質量%以上,更好是0.2質量%以上,進一步更好是0.3質量%以上。對上限無特別限定,可以是較好為10質量%以下,更好為5質量%以下,進一步更好為3質量%以下等。
-(e)固化促進劑-
作為固化促進劑,可例舉例如磷類固化促進劑、胺類固化促進劑、咪唑類固化促進劑、胍類固化促進劑、金屬類固化促進劑、有機過氧化物類固化促進劑等,較好是磷類固化促進劑、胺類固化促進劑、咪唑類固化促進劑、金屬類固化促進劑,更好是胺類固化促進劑、咪唑類固化促進劑、金屬類固化促進劑。固化促進劑可單獨使用1種,也可2種以上組合使用。此外,固化促進劑可使用市售品。
作為磷類固化促進劑,可例舉例如三苯基膦、鏻硼酸鹽化合物、四苯基鏻四苯基硼酸鹽、正丁基鏻四苯基硼酸鹽、四丁基鏻癸酸鹽、(4-甲基苯基)三苯基鏻硫氰酸鹽、四苯基鏻硫氰酸鹽、丁基三苯基鏻硫氰酸鹽等,較好是三苯基膦、四丁基鏻癸酸鹽。
作為胺類固化促進劑,可例舉例如三乙胺、三丁胺等三烷基胺、4-二甲基氨基吡啶、芐基二甲胺、2,4,6-三(二甲基氨基甲基)苯酚、1,8-二氮雜雙環[5.4.0]十一烯等,較好是4-二甲基氨基吡啶、1,8-二氮雜雙環[5.4.0]十一烯。
作為咪唑類固化促進劑,可例舉例如2-甲基咪唑、2-十一烷基咪唑、2-十七烷基咪唑、1,2-二甲基咪唑、2-乙基-4-甲基咪唑、1,2-二甲基咪唑、2-乙基-4-甲基咪唑、2-苯基咪唑、2-苯基-4-甲基咪唑、1-芐基-2-甲基咪唑、1-芐基-2-苯基咪唑、1-氰基乙基-2-甲基咪唑、1-氰基乙基-2-十一烷基咪唑、1-氰基乙基-2-乙基-4-甲基咪唑、1-氰基乙基-2-苯基咪唑、1-氰基乙基-2-十一烷基咪唑鎓偏苯三酸鹽、1-氰基乙基-2-苯基咪唑鎓偏苯三酸鹽、2,4-二氨基-6-[2'-甲基咪唑基-(1')]-乙基-均三嗪、2,4-二氨基-6-[2'-十一烷基咪唑基-(1')]-乙基-均三嗪、2,4-二氨基-6-[2'-乙基-4'-甲基咪唑基-(1')]-乙基-均三嗪、2,4-二氨基-6-[2'-甲基咪唑基-(1')]-乙基-均三嗪異氰脲酸加成物、2-苯基咪唑異氰脲酸加成物、2-苯基-4,5-二羥基甲基咪唑、2-苯基-4-甲基-5-羥基甲基咪唑、2,3-二氫-1h-吡咯并[1,2-a]苯并咪唑、1-十二烷基-2-甲基-3-芐基咪唑鎓氯化物、2-甲基咪唑啉、2-苯基咪唑啉等咪唑化合物及咪唑化合物與環氧樹脂的加合物,較好是2-乙基-4-甲基咪唑、1-芐基-2-苯基咪唑。
作為咪唑類固化促進劑,可使用市售品,可例舉例如三菱化學株式會社制的“p200-h50”等。
作為胍類固化促進劑,可例舉例如雙氰胺、1-甲基胍、1-乙基胍、1-環己基胍、1-苯基胍、1-(鄰甲苯基)胍、二甲基胍、二苯基胍、三甲基胍、四甲基胍、五甲基胍、1,5,7-三氮雜雙環[4.4.0]癸-5-烯、7-甲基-1,5,7-三氮雜雙環[4.4.0]癸-5-烯、1-甲基雙胍、1-乙基雙胍、1-正丁基雙胍、1-正十八烷基雙胍、1,1-二甲基雙胍、1,1-二乙基雙胍、1-環己基雙胍、1-烯丙基雙胍、1-苯基雙胍、1-(鄰甲苯基)雙胍等,較好是雙氰胺、1,5,7-三氮雜雙環[4.4.0]癸-5-烯。
作為金屬類固化促進劑,可例舉例如鈷、銅、鋅、鐵、鎳、錳、錫等金屬的有機金屬絡合物或有機金屬鹽。作為有機金屬絡合物的具體例子,可例舉乙酰丙酮合鈷(ii)、乙酰丙酮合鈷(iii)等有機鈷絡合物、乙酰丙酮合銅(ii)等有機銅絡合物、乙酰丙酮合鋅(ii)等有機鋅絡合物、乙酰丙酮合鐵(iii)等有機鐵絡合物、乙酰丙酮合鎳(ii)等有機鎳絡合物、乙酰丙酮合錳(ii)等有機錳絡合物等。作為有機金屬鹽,可例舉例如辛酸鋅、辛酸錫、環烷酸鋅、環烷酸鈷、硬脂酸錫、硬脂酸鋅等。
作為有機過氧化物類固化促進劑,可例舉例如過氧化二異丙苯、過氧化環己酮、過氧化苯甲酸叔丁酯、過氧化甲基乙基酮、過氧化二異丙苯、過氧化叔丁基異丙苯、過氧化二叔丁基、過氧化氫二異丙苯、氫過氧化枯烯、過氧化氫叔丁基等。作為有機過氧化物類固化促進劑,可使用市售品,可例舉例如日油株式會社制的“percumyld”等。
樹脂組合物中的固化促進劑的含量無特別限定,將環氧樹脂和固化劑的不揮發成分設為100質量%時,較好是0.005質量%~3質量%。
-(f)阻燃劑-
作為阻燃劑,可例舉例如有機磷類阻燃劑、有機類含氮的磷化合物、氮化合物、有機硅類阻燃劑、金屬氫氧化物等。阻燃劑可單獨使用1種,也可2種以上并用。
作為阻燃劑,可使用市售品,可例舉例如三光株式會社制的“hca-hq”、大八化學工業株式會社制的“px-200”等。
樹脂組合物包含阻燃劑的情況下,阻燃劑的含量無特別限定,但較好是0.5質量%~20質量%,更好是0.5質量%~15質量%,進一步更好是0.5質量%~10質量%。
-(g)有機填充材料-
從使伸長率提高的觀點來看,樹脂組合物可包含(g)有機填充材料。作為有機填充材料,可使用形成印刷布線板的絕緣層時能使用的任意的有機填充材料,可例舉例如橡膠粒子、聚酰胺微粒、有機硅粒子等。
作為橡膠粒子,可使用市售品,可例舉例如陶氏化學日本株式會社制的“exl2655”、愛克(aica)工業株式會社制的“ac3816n”等。
樹脂組合物包含有機填充材料的情況下,有機填充材料的含量無特別限定,但較好是0.1質量%~20質量%,更好是0.2質量%~10質量%,進一步更好是0.3質量%~5質量%或0.5質量%~3質量%。
-(h)其他添加劑-
樹脂組合物還可根據需要包含其他添加劑,作為這樣的其他添加劑,可例舉例如有機銅化合物、有機鋅化合物和有機鈷化合物等有機金屬化合物以及增稠劑、消泡劑、均化劑、密合性賦予劑和著色劑等樹脂添加劑等。
此外,從制造柔性印刷布線板的觀點來看,樹脂組合物較好是還包含分子內具有聚丁二烯結構、氨基甲酸酯結構、酰亞胺結構且分子末端具有酚結構的聚酰亞胺樹脂。包含該聚酰亞胺樹脂的情況下,含量較好是10質量%~85質量%,更好是12質量%~50質量%,進一步更好是15質量%~30質量%。
該聚酰亞胺的詳細內容可參考國際公開第2008/153208號的記載,該內容引用至本說明書。
從抑制固化不均和波動的產生的觀點來看,第一實施方式的粘接片中的120℃以上時的樹脂組合物層的最低熔融粘度y較好是1000泊以上,更好是1500泊以上,進一步更好是2000泊以上、或2500泊以上、或4000泊以上。上限較好是1000000泊以下,更好是500000泊以下,進一步更好是400000泊以下、或50000泊以下、或40000泊以下。
第二實施方式的粘接片中的t1(℃)以上時的樹脂組合物層的最低熔融粘度是4000泊以上,較好是4500泊以上,更好是5000泊以上,進一步更好是6000泊以上。上限較好是1000000泊以下,更好是600000泊以下,進一步更好是100000泊以下、50000泊以下或10000泊以下。通過使該最低熔融粘度在4000泊以上,可抑制固化不均和波動的產生。
在此,樹脂組合物層的“最低熔融粘度”是指樹脂組合物層的樹脂熔融時樹脂組合物層呈現的最低粘度。具體來說,如果以一定的升溫速度加熱樹脂組合物層使樹脂熔融,則初期階段熔融粘度隨著溫度上升而下降,然后超過一定溫度后熔融粘度隨著溫度上升而上升。“最低熔融粘度”是指上述極小點的熔融粘度。樹脂組合物層的最低熔融粘度及最低熔融粘度溫度可通過動態粘彈性法進行測定。
樹脂組合物層的厚度無特別限定,但從印刷布線板的薄型化的觀點來看,較好是5μm~100μm,更好是10μm~90μm,進一步更好是15μm~80μm。
本發明的粘接片也包括同時滿足第一和第二實施方式的形態的粘接片,較好是該形態的粘接片。同時滿足第一和第二實施方式的形態是指支撐體的x((eamd-ebmd)+(eatd-ebtd))為4以下,t1(℃)以上時的樹脂組合物層的最低熔融粘度為4000泊以上,且120℃以上時的樹脂組合物層的最低熔融粘度y滿足y>2700x的粘接片。
[印刷布線板的制造方法]
對于本發明的第一實施方式的印刷布線板的制造方法而言,該方法是包括下述工序的印刷布線板的制造方法:(a)準備具備支撐體和設置于該支撐體上的樹脂組合物層的粘接片的工序、(b)以樹脂組合物層與內層基板接合的方式將粘接片疊層于內層基板的工序、以及(c)通過將粘接片從t1(℃)加熱至t2(℃)而進行熱固化,形成絕緣層的工序,其中,將(eamd-ebmd)與(eatd-ebtd)之和((eamd-ebmd)+(eatd-ebtd))設為x、將120℃以上時的樹脂組合物層的最低熔融粘度設為y(泊)時,以滿足y>2700x的關系的方式進行熱固化,所述(eamd-ebmd)是從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebmd(%)之差,所述(eatd-ebtd)是從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebtd(%)之差。
對于本發明的第二實施方式的印刷布線板的制造方法而言,該方法是包括下述工序的印刷布線板的制造方法:(a)準備具備支撐體和設置于該支撐體上的樹脂組合物層的粘接片的工序、(b)以樹脂組合物層與內層基板接合的方式將粘接片疊層于內層基板的工序、以及(c)通過將粘接片從t1(℃)加熱至t2(℃)而進行熱固化,形成絕緣層的工序,其中,以(eamd-ebmd)與(eatd-ebtd)之和((eamd-ebmd)+(eatd-ebtd))為4以下、t1(℃)以上時的樹脂組合物層的最低熔融粘度為4000泊以上的方式進行熱固化,所述(eamd-ebmd)是從t1(℃)加熱至t2(℃)時的md方向的支撐體的最大膨脹系數eamd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebmd(%)之差,所述(eatd-ebtd)是從t1(℃)加熱至t2(℃)時的td方向的支撐體的最大膨脹系數eatd(%)與加熱結束的時刻的t2(℃)時的支撐體的膨脹系數ebtd(%)之差。
以下,對第一和第二實施方式的各工序進行說明。
<工序(a)>
工序(a)中,準備具備支撐體以及設置于該支撐體上的樹脂組合物層的粘接片。
粘接片如上述[粘接片]中所說明。第一實施方式的印刷布線板的制造方法的情況下,較好是準備第一實施方式的粘接片,第二實施方式的印刷布線板的制造方法的情況下,較好是準備第二實施方式的粘接片。
粘接片例如可如下制造:制備在有機溶劑中溶解樹脂組合物而得的樹脂清漆,用金屬型涂布機(diecoater)等將該樹脂清漆涂布于支撐體上,使其干燥而在支撐體上形成樹脂組合物層。
作為有機溶劑,可例舉例如丙酮、甲基乙基酮和環己酮等酮類;乙酸乙酯、乙酸丁酯、乙酸溶纖劑、丙二醇單甲醚乙酸酯和卡必醇乙酸酯等乙酸酯類;溶纖劑和丁基卡必醇等卡必醇類;甲苯和二甲苯等芳香族烴類;二甲基甲酰胺、二甲基乙酰胺和n-甲基吡咯烷酮等酰胺類溶劑等。有機溶劑可單獨使用1種,也可2種以上并用。
干燥可通過加熱、熱風吹拂等公知的干燥方法實施。干燥條件無特別限定,以樹脂組合物層中的有機溶劑的含量(殘留溶劑量)達到10質量%以下、較好是5質量%以下的條件進行干燥。此外,從使樹脂組合物層的操作性提高、防止制成粘接片時的熔融粘度的上升的觀點來看,殘留溶劑量較好是0.5質量%以上,更好是1質量%以上。根據樹脂清漆中的有機溶劑的沸點而不同,例如使用含30質量%~60質量%的有機溶劑的樹脂清漆的情況下,可通過在50℃~150℃干燥3分鐘~10分鐘來形成樹脂組合物層。
粘接片中,可在樹脂組合物層的不與支撐體接合的表面(即,與支撐體相反的一側的面)進而疊層保護膜,且該保護膜為與已經說明過的支撐體同樣的膜。保護膜的厚度無特別限定,例如為1μm~40μm。通過疊層保護膜,可防止樹脂組合物層的表面附著垃圾等或形成損傷。粘接片可卷取成卷狀保存,制造印刷布線板時,可通過剝離保護膜來使用。
<工序(b)>
工序(b)中,以樹脂組合物層與內層基板接合的方式將粘接片疊層于內層基板。
工序(b)中使用的“內層基板”主要是指玻璃環氧基板、金屬基板、聚酯基板、聚酰亞胺基板、bt樹脂基板、熱固化型聚苯醚基板等基板、或者在該基板的單面或兩面形成有圖案加工而成的導體層(電路)的電路基板。此外,作為制造印刷布線板時需進一步形成絕緣層和/或導體層的已形成有1層以上的絕緣層和/或導體層的中間制造物的疊層結構體也包含在本發明中所說的“內層基板”中。
粘接片與內層基板的疊層(接合)例如可通過自支撐體側將粘接片加熱壓接于內層基板來進行。作為將粘接片加熱壓接于內層基板的構件(以下也稱“加熱壓接構件”),可例舉例如加熱后的金屬板(不銹鋼(sus)端面板等)或金屬輥(sus輥)等。應予說明,較好是不使加熱壓接構件直接與粘接片接觸來進行壓制,而是隔著由耐熱橡膠等彈性材料形成的片材等進行壓制,使得粘接片充分順應內層基板的表面凹凸。
加熱壓接時的溫度較好是在80℃~160℃、更好是90℃~140℃、進一步更好是100℃~120℃的范圍內,加熱壓接時的壓力較好是在0.098mpa~1.77mpa、更好是0.29mpa~1.47mpa的范圍內,加熱壓接時的時間較好是在20秒~400秒、更好是30秒~300秒的范圍內。粘接片與內層基板的接合較好是在壓力26.7hpa以下的減壓條件下實施。
粘接片與內層基板的接合可通過市售的真空層合機進行。作為市售的真空層合機,可例舉例如株式會社名機制作所制的真空加壓式層合機、nikko-materials株式會社制的真空敷料器等。
可在粘接片與內層基板的接合后,常壓下(大氣壓下),例如將加熱壓接構件自支撐體側壓制來進行疊層的粘接片的平滑化處理。平滑化處理的壓制條件可采用與上述疊層的加熱壓接條件同樣的條件。平滑化處理可通過市售的層合機進行。應予說明,疊層和平滑化處理可使用上述的市售的真空層合機連續進行。
<工序(c)>
工序(c)中,通過將粘接片從t1(℃)加熱至t2(℃)而使其熱固化,形成絕緣層。工序(c)中,只要可通過調整熱固化時的加熱條件來控制支撐體的膨脹、收縮,從t1(℃)加熱至t2(℃)的過程無特別限定,可以是采用1步的熱固化,也可以是采用2步的熱固化。
熱固化的條件考慮所選擇的支撐體的特性和樹脂組合物層的特性決定,可適用形成印刷布線板的絕緣層時通常所采用的條件。
例如,樹脂組合物層的熱固化條件根據樹脂組合物層的組成而不同,固化溫度可設在50℃~240℃或150℃~240℃的范圍內(較好是155℃~230℃,更好是160℃~220℃,進一步更好是170℃~210℃,再進一步更好是180℃~200℃)。固化時間可設在5分鐘~100分鐘的范圍內(較好是10分鐘~80分鐘,更好是10分鐘~50分鐘)。此外,固化條件可考慮樹脂組合物層通過熔融以自調整的方式平坦化的條件決定。熱固化可在常壓下、減壓下、加壓下中的任一條件下實施。
優選的一個實施方式中,工序(c)中,將粘接片在t1(℃)加熱后,再在t2(℃)加熱來進行熱固化。即,工序(c)較好是包括
i)將樹脂組合物層在溫度t1(℃)(其中,50℃≤t1≤150℃)下進行加熱的工序、和
ii)將加熱后的樹脂組合物層在溫度t2(℃)(其中,150℃≤t2≤240℃)下進行加熱的工序,
即以2步進行加熱。
上述i)的加熱中,作為t1(℃),也根據樹脂組合物層的組成而不同,但較好是滿足50℃≤t1≤150℃的關系,更好是滿足80℃≤t1≤150℃的關系,進一步更好是滿足120℃≤t1≤150℃的關系。
可在t1(℃)下加熱后,保持t1(℃)一定時間。在t1(℃)保持的時間也根據t1的值而不同,但較好是10分鐘~150分鐘,更好是15分鐘~60分鐘,進一步更好是20分鐘~40分鐘。
上述i)的加熱可在常壓下實施,也可在減壓下實施,還可在加壓下實施,但較好是在0.075mmhg~3751mmhg(0.1hpa~5000hpa)的范圍內、更好是在1mmhg~1875mmhg(1.3hpa~2500hpa)的范圍內的壓力下實施。
上述ii)的熱固化中,溫度t2(℃)也根據樹脂組合物層的組成而不同,但較好是滿足150℃≤t2≤240℃的關系,更好是滿足155℃≤t2≤200℃的關系,進一步更好是滿足170℃≤t2≤180℃的關系。
在t2(℃)下熱固化的時間也根據t2的值而不同,但較好是5分鐘~100分鐘,更好是10分鐘~80分鐘,進一步更好是10分鐘~50分鐘。
上述ii)的熱固化可在常壓下實施,也可在減壓下實施,還可在加壓下實施。較好是在與上述i)同樣的壓力下實施。
應予說明,t1(℃)和t2(℃)較好是滿足10℃≤t2-t1≤150℃的關系,更好是滿足15℃≤t2-t1≤140℃的關系,進一步更好是滿足15℃≤t2-t1≤120℃的關系,特別好是滿足15℃≤t2-t1≤100℃的關系。
可在上述i)的加熱后,讓樹脂組合物層一度散熱,再實施上述ii)的熱固化。或者,可在上述i)的加熱后,在沒有讓樹脂組合物層散熱的情況下,實施上述ii)的熱固化。優選的一個實施方式中,工序(c)在上述i)的加熱與上述ii)的熱固化之間還包括從t1(℃)升溫至t2(℃)的工序。所述實施方式中,從t1(℃)至t2(℃)的升溫速度較好是1.5℃/分鐘~30℃/分鐘,更好是2℃/分鐘~30℃/分鐘,進一步更好是4℃/分鐘~20℃/分鐘,再進一步更好是4℃/分鐘~10℃/分鐘。應予說明,可在所述升溫的過程中開始樹脂組合物層的熱固化。
應予說明,通過采用1步的加熱進行熱固化時,作為t1(℃),也根據樹脂組合物層的組成而不同,但較好是滿足0℃≤t1≤50℃的關系,更好是滿足5℃≤t1≤40℃的關系,進一步更好是滿足10℃≤t1≤30℃的關系。
絕緣層的厚度與樹脂組合物層的厚度相同,優選的范圍也相同。
<工序(d)>
可在工序(c)結束后,進行除去支撐體的工序(d)。
對于支撐體的除去,可通過目前公知的任意合適的方法剝離除去,也可通過自動剝離裝置以機械方式剝離除去。通過所述工序(d)的實施,所形成的絕緣層的表面露出。
制造印刷布線板時,還可實施(e)在絕緣層開孔的工序、(f)對絕緣層進行粗糙化處理的工序、(g)在絕緣層表面形成導體層的工序。這些工序(e)~工序(g)可按照印刷布線板的制造所用的本領域的技術人員公知的各種方法實施。
工序(e)是在絕緣層開孔的工序,可籍此在絕緣層形成通孔、透孔。開孔的工序可使用例如鉆孔機、激光器(二氧化碳激光器、yag激光器等)、等離子體等來實施。應予說明,工序(e)可在工序(c)與工序(d)之間實施,也可在工序(d)之后實施。
工序(f)是對絕緣層進行粗糙化處理的工序。所述粗糙化處理的步驟、條件無特別限定,可采用形成印刷布線板的絕緣層時通常所使用的公知的步驟、條件。工序(f)例如可以是如下工序:依次實施采用膨潤液的膨潤處理、采用氧化劑的粗糙化處理、采用中和液的中和處理來對絕緣層進行粗糙化處理。膨潤液無特別限定,可例舉堿溶液、表面活性劑溶液等,較好是堿溶液,作為該堿溶液,更好是氫氧化鈉溶液、氫氧化鉀溶液。作為市售的膨潤液,可例舉例如atotechjapan株式會社制的“swellingdipsecuriganthp”、“swellingdipsecuriganthsbu”等。采用膨潤液的膨潤處理無特別限定,例如可通過將絕緣層在30℃~90℃的膨潤液中浸漬1分鐘~20分鐘來進行。作為氧化劑,無特別限定,可例舉例如在氫氧化鈉的水溶液中溶解高錳酸鉀或高錳酸鈉而得的堿性高錳酸溶液。采用堿性高錳酸溶液等氧化劑的粗糙化處理較好是使絕緣層在加熱至60℃~80℃的氧化劑溶液中浸漬10分鐘~30分鐘來進行。此外,堿性高錳酸溶液中的高錳酸鹽的濃度較好是5質量%~10質量%。作為市售的氧化劑,可例舉例如atotechjapan株式會社制的“concentratecompactcp”、“dosingsolutionsecurighanthp”等堿性高錳酸溶液。此外,作為中和液,較好是酸性的水溶液,作為市售品,可例舉例如atotechjapan株式會社制的“reductionsolutionsecuriganthp”等。采用中和液的處理可通過使進行了采用氧化劑溶液的粗糙化處理的處理面在30℃~80℃的中和液中浸漬5分鐘~30分鐘來進行。
工序(g)是在絕緣層的表面形成導體層的工序。
用于導體層的導體材料無特別限定。優選的實施方式中,導體層包含選自金、鉑、鈀、銀、銅、鋁、鈷、鉻、鋅、鎳、鈦、鎢、鐵、錫和銦的1種以上的金屬。導體層可以是單金屬層或合金層,作為合金層,可例舉例如由選自上述金屬的2種以上的金屬的合金(例如鎳-鉻合金、銅-鎳合金和銅-鈦合金)形成的層。其中,從導體層形成的難易度、成本、圖案加工的難易度等觀點來看,較好是鉻、鎳、鈦、鋁、鋅、金、鈀、銀或銅的單金屬層或者鎳-鉻合金、銅-鎳合金、銅-鈦合金的合金層,更好是鉻、鎳、鈦、鋁、鋅、金、鈀、銀或銅的單金屬層或者鎳-鉻合金的合金層,更好是銅的單金屬層。
導體層可以是單層結構,也可以是由不同種類的金屬或合金形成的單金屬層或者合金層疊層2層以上而得的多層結構。導體層為多層結構的情況下,與絕緣層相接的層較好是鉻、鋅或鈦的單金屬層或者鎳-鉻合金的合金層。
導體層的厚度根據所期望的印刷布線板的設計而不同,但一般為3μm~35μm,較好是5μm~30μm。
導體層可通過鍍覆形成。例如,可通過半添加法、全添加法等目前公知的技術在絕緣層的表面進行鍍覆,形成具有所期望的布線圖案的導體層。以下,示出通過半添加法形成導體層的例子。
首先,在絕緣層的表面通過無電解鍍覆形成鍍覆晶種層(めっきシード層)。接著,在形成的鍍覆晶種層上對應所期望的布線圖案而形成使鍍覆晶種層的一部分露出的掩模圖案。在露出的鍍覆晶種層上通過電解鍍覆形成金屬層后,除去掩模圖案。然后,通過蝕刻等除去不需要的鍍覆晶種層,從而可形成具有所期望的布線圖案的導體層。
通過本發明的印刷布線板的制造方法制造的印刷布線板可抑制絕緣層的波動等。即,帶來絕緣層的厚度的偏差被抑制了的印刷布線板。波動較好是1μm以下,更好是0.8μm以下,進一步更好是0.5μm以下。偏差的下限無特別限定,可設為0.01μm以上等。波動的測定可按照后述的[波動的測定]中記載的方法進行測定。
通過本發明的印刷布線板的制造方法制造的印刷布線板通過肉眼觀察發現絕緣層形成后的固化不均得到抑制。即,帶來固化不均被抑制了的印刷布線板。固化不均的測定可按照后述的[絕緣層表面的評價]中記載的方法進行測定。
[半導體裝置]
可使用通過本發明的制造方法得到的印刷布線板,制造具備所述印刷布線板的半導體裝置。
作為半導體裝置,可例舉供于電氣制品(例如電腦、手機、數碼相機和電視機等)和交通工具(例如二輪機動車、汽車、電車、船舶和航空器等)等的各種半導體裝置。
本發明的半導體裝置可通過將部件(半導體芯片)安裝于印刷布線板的導通位置來制造。“導通位置”是指“印刷布線板中傳導電信號的位置”,其位置可以在表面,也可以是被包埋的位置。此外,半導體芯片只要是以半導體為材料的電氣電路元件即可,無特別限定。
制造本發明的半導體裝置時的半導體芯片的安裝方法只要半導體芯片有效地發揮作用即可,無特別限定,具體可例舉引線接合安裝方法、倒裝芯片安裝方法、采用無凸點疊層(bumplessbuild-uplayer,bbul)的安裝方法、采用各向異性導電膜(acf)的安裝方法、采用非導電性膜(ncf)的安裝方法等。在此,“采用無凸點疊層(bbul)的安裝方法”是指“將半導體芯片直接埋入印刷布線板的凹部,使半導體芯片與印刷布線板上的布線連接的安裝方法”。
[疊層結構體]
本發明的疊層結構體是具有內層基板、設置于該內層基板的絕緣層、以及接合于該絕緣層的支撐體的疊層結構體,其中,在從支撐體的端部相對于支撐體的尺寸除去5%后的范圍內,該絕緣層的厚度的偏差在1μm以下。
參照圖3和圖4對本發明的疊層結構體的構成例進行說明。圖3是疊層結構體的示意性俯視圖。圖4是示出將疊層結構體在圖3中的iii-iii點劃線的位置截斷而得的端面的示意圖。
如圖3和圖4中所示的一例,疊層結構體10具有內層基板30、設置于該內層基板30的絕緣層22、以及接合于該絕緣層22的支撐體21。
應予說明,該疊層結構體10中,形成于內層基板30的絕緣層22是指已經說明了的通過粘接片20新形成的絕緣層。疊層結構體10中的絕緣層22不僅限于1層,還可包括在內層基板30的一側的表面側疊層2層以上的絕緣層22的形態。此外,疊層結構體10具有2層以上的絕緣層的情況下,2層以上的絕緣層中的至少1層通過下述工序形成即可:將具有已經說明了的特性的具備支撐體21的粘接片疊層于內層基板30后,將樹脂組合物層熱固化后,剝離除去支撐體21。
疊層結構體10中,在從疊層結構體10的支撐體的端部相對于支撐體的尺寸除去5%后的范圍內的絕緣層的厚度的偏差在1μm以下,較好是0.8μm以下,更好是0.5μm以下。偏差的下限無特別限定,可設為0.01μm以上等。
如圖3和圖4中所示的一例,這里所說的“從支撐體的端部相對于支撐體的尺寸除去5%后的范圍”是指疊層結構體10的除去端部10b后的范圍。具體是指相對于支撐體的尺寸(支撐體的長度)10a除去了兩端部10b后的范圍(中央部10c的范圍),端部10b是相對于支撐體的尺寸10a為2.5%的長度。圖3和圖4中,僅md方向示出支撐體的尺寸(支撐體的長度)10a,但對于td方向也與md方向同樣。
實施例
以下,通過實施例對本發明進行具體說明。本發明并不局限于這些實施例。應予說明,以下中,只要沒有另行明示,“份”和“%”分別表示“質量份”和“質量%”。
[實施例1]
<粘接片的制造>
-支撐體的預加熱條件-
下述的實施例和比較例中制備支撐體時采用的預加熱條件示于下表。應予說明,表中的“張力”是指施加于支撐體的td方向的張力。
[表1]
(1)支撐體的制備
將帶醇酸樹脂類脫模層的pet膜(琳得科株式會社制“al5”,厚38μm,以下也稱“脫模pet膜”)在空氣氣氛中于常壓下以所述表的預加熱條件2對pet的td方向施加張力的同時進行加熱,得到支撐體。
(2)樹脂清漆a的制備
一邊攪拌一邊使30份聯苯型環氧樹脂(環氧當量約290,日本化藥株式會社制“nc3000h”)、5份萘型四官能環氧樹脂(環氧當量162,dic株式會社制“hp-4700”)、15份液態雙酚a型環氧樹脂(環氧當量180,三菱化學株式會社制“jer828el”)和2份苯氧樹脂(重均分子量35000,三菱化學株式會社制“yl7553bh30”,固體成分30質量%的甲基乙基酮(mek)溶液)加熱溶解于8份mek和8份環己酮的混合溶劑。向其中混合32份含三嗪骨架的苯酚酚醛類固化劑(酚式羥基當量約124,dic株式會社制“la-7054”,不揮發成分60質量%的mek溶液)、0.2份磷類固化促進劑(北興化學工業株式會社制“tbp-da”,四丁基鏻癸酸鹽)、160份通過氨基硅烷類偶聯劑(信越化學株式會社制“kbm573”)進行了表面處理的球狀二氧化硅(株式會社admatechs制“soc2”,平均粒徑0.5μm)、2份聚乙烯醇縮丁醛樹脂溶液(重均分子量27000,玻璃化轉變溫度105℃,積水化學工業株式會社制“ks-1”,不揮發成分15質量%的乙醇和甲苯的質量比為1:1的混合溶液),通過高速旋轉混合機均勻分散,制成樹脂清漆。將樹脂清漆中的不揮發成分的合計質量設為100質量%時,無機填充材料(球狀二氧化硅)的含量為69.5質量%。
(3)粘接片的制造
將上述(2)中制成的樹脂清漆a通過金屬型涂布機均勻地涂布于上述(1)中制成的支撐體的脫模層上,在80~120℃(平均100℃)干燥6分鐘而形成樹脂組合物層。所得的樹脂組合物層的厚度為40μm,殘留溶劑量為約2質量%。接著,在樹脂組合物層上粘合作為保護膜的聚丙烯膜(厚15μm)的同時卷取成卷狀。將所得的卷狀的粘接片切成寬507mm,獲得尺寸507mm×336mm的粘接片1。
<評價、測定用樣品的制備>
(1)內層電路基板的準備
將玻璃布基材環氧樹脂兩面覆銅疊層板(銅箔厚度18μm,基板厚度0.8mm,松下電工株式會社制“r1515a”)的兩面浸漬于美格(mec)株式會社制“cz8100”進行銅表面的粗糙化處理。
(2)粘接片的疊層
將制成的粘接片1使用分批式真空加壓層合機(株式會社名機制作所制“mvlp-500”)以樹脂組合物層與內層電路基板接合的方式在內層電路基板的兩面進行層合處理。層合處理如下進行:減壓30秒使氣壓達到13hpa以下后,以100℃、壓力0.74mpa壓接30秒。應予說明,粘接片1在剝離保護膜后供于疊層。
(3)樹脂組合物層的熱固化
粘接片1的疊層后,在帶有支撐體的狀態下,以下表中記載的包括溫度t1(℃)、溫度t2(℃)的2階段的加熱條件(即圖1的形態)中的加熱條件2將樹脂組合物層熱固化而形成絕緣層。應予說明,加熱的開始溫度為20℃(室溫),到溫度t1為止的升溫速度設為8℃/分鐘。所得的絕緣層的內層電路上的厚度為40μm。
[表2]
[實施例2]
實施例1中,除了將樹脂組合物層的加熱條件2改為加熱條件3以外,與實施例1同樣地進行操作,制成評價、測定用樣品。
[實施例3]
實施例1中,除了將樹脂清漆a改為以下的樹脂清漆b,加熱條件2改為加熱條件5,支撐體的預加熱條件2改為預加熱條件1以外,與實施例1同樣地進行操作,制成評價、測定用樣品。
-樹脂清漆b的制備-
一邊攪拌一邊使28份液態雙酚a型環氧樹脂(環氧當量180,三菱化學株式會社制“jer828el”)、28份萘型四官能環氧樹脂(環氧當量162,dic株式會社制“hp-4700”)加熱溶解于15份甲基乙基酮(以下簡稱“mek”)和15份環己酮的混合溶劑。向其中混合110份具有酚醛結構的萘酚類固化劑(酚式羥基當量215,新日鐵住金化學株式會社制“sn485”,固體成分50質量%的mek溶液)、0.1份固化促進劑(四國化成工業株式會社制“2e4mz”,2-苯基-4-甲基咪唑)、70份球形二氧化硅(株式會社admatechs制“so-c2”,平均粒徑0.5μm)、35份聚乙烯醇縮丁醛樹脂溶液(積水化學工業株式會社制“ks-1”,重均分子量27000,玻璃化轉變溫度105℃,固體成分15%的乙醇和甲苯的1:1的混合溶液),通過高速旋轉混合機均勻分散,制成樹脂清漆。將樹脂清漆中的不揮發成分設為100質量%時,樹脂清漆中的無機填充材料的含量為38質量%。此外,[環氧樹脂的環氧基的總數]:[固化劑的反應基團的總數]=1:0.78。
[實施例4]
實施例3中,除了將加熱條件5改為加熱條件6以外,與實施例3同樣地進行操作,制成評價、測定用樣品。
[實施例5]
實施例3中,除了將加熱條件5改為加熱條件7以外,與實施例3同樣地進行操作,制成評價、測定用樣品。
[實施例6]
實施例3中,除了將加熱條件5改為加熱條件8以外,與實施例3同樣地進行操作,制成評價、測定用樣品。
[實施例7]
實施例3中,除了將加熱條件5改為加熱條件4以外,與實施例3同樣地進行操作,制成評價、測定用樣品。
[比較例1]
實施例1中,除了將加熱條件2改為加熱條件1以外,與實施例1同樣地進行操作,制成評價、測定用樣品。
[比較例2]
實施例3中,除了將加熱條件5改為加熱條件6,支撐體的預加熱條件1改為預加熱條件3以外,與實施例3同樣地進行操作,制成評價、測定用樣品。
[支撐體的膨脹系數的測定]
<支撐體的td方向的膨脹系數、收縮率的測定>
以實施例和比較例中制成的支撐體的td方向為沿長邊的方向的方式切出尺寸20mm×4mm的長方形的試驗片。對于該試驗片,使用熱機械分析裝置(seikoinstruments株式會社制“tma-ss6100”),在大氣氣氛下,以9.8mmn荷重按壓的同時,在上表中記載的包括溫度t1(℃)、溫度t2(℃)的2階段的加熱條件下,對加熱處理的全過程測定膨脹系數,求出從溫度t1(℃)加熱至溫度t2(℃)時的td方向的最大膨脹系數eatd(%)、及加熱結束的時刻的膨脹系數ebtd(%)。
對實施例和比較例中制成的支撐體,求出上述最大膨脹系數eatd與膨脹系數ebtd之差(eatd-ebtd)。
<支撐體的md方向的膨脹系數、收縮率的測定>
以實施例和比較例中制成的支撐體的md方向為長邊側切出尺寸20mm×4mm的試驗片。對于該試驗片,與上述同樣地進行操作,求出從溫度t1(℃)加熱至溫度t2(℃)時的md方向的最大膨脹系數eamd(%)、及加熱結束的時刻的膨脹系數ebmd(%)。
對實施例和比較例中制成的支撐體,求出最大膨脹系數eamd與膨脹系數ebmd之差(eamd-ebmd)。此外,將(eamd-ebmd)與(eatd-ebtd)相加,算出支撐體的位移量x。
[最低熔融粘度的測定]
對于實施例和比較例中制成的粘接片的樹脂組合物層,使用動態粘彈性測定裝置(株式會社ubm制“rheosol-g3000”)測定熔融粘度。將1g樹脂組合物作為試樣,使用直徑18mm的平行板,測定開始溫度設為60℃,升溫速度設為8℃/分鐘,在上表中記載的包括溫度t1(℃)、溫度t2(℃)的2階段的加熱條件下,以測定溫度間隔2.5℃、頻率1hz、變形1deg的測定條件測定動態粘彈性模量。確認溫度t1(℃)以上時的最低粘度(泊)。此外,確認120℃以上的最低熔融粘度y(泊)。
[波動的測定]
對于實施例和比較例中制成的評價、測定用樣品中的絕緣層的表面,使用非接觸型表面粗糙度計(veecoinstrumentsinc.制“wykogt-x3”),在vsi接觸模式下通過50倍透鏡將測定區域設為121μm×92μm進行測定,根據所得的數值(絕緣層的最大高度與最小高度之差)求出波動(μm)。對于各絕緣層,在產生固化不均的情況下,對產生固化不均的部分測定3處(其中,從絕緣層的端部相對于絕緣層的尺寸除去了5%的范圍),在未產生固化不均的情況下,測定從絕緣層的端部相對于絕緣層的尺寸除去了5%的范圍后隨機選擇的3處。
[絕緣層表面的評價]
絕緣層表面的評價通過采用加熱處理后的肉眼觀察和上述的波動測定的綜合評價來進行。
在從支撐體的端部除去5%后的范圍內滿足以下所有條件的情況判定為“○”,滿足任一項的情況判定為“△”,及兩者均不滿足的情況判定為“×”,
(條件)
·利用肉眼觀察在絕緣層形成后未產生固化不均;
·在絕緣層形成后未產生超過1.0μm的波動。
[表3]
符號的說明
10疊層結構體
10a支撐體的尺寸(支撐體的長度)
10b端部
10c中央部
20粘接片
21支撐體
22絕緣層
30內層基板。
- 還沒有人留言評論。精彩留言會獲得點贊!