一種球形摻雜錳酸鋰的制備方法與流程
本發明涉及一種球形摻雜錳酸鋰的制備方法,屬于鋰離子電池正極材料領域。
背景技術:
鋰離子電池具有工作電壓高、比能量高、無記憶效應等特點,在消費類數碼產品(智能手機、筆記本電腦等)、電動汽車及儲能電池等領域得到應用。近年來,隨著鋰離子電池技術不斷進步,電動汽車快速發展,其中teslamodels、日產leaf、雪佛蘭volt、比亞迪e6等電動車獲得了不錯的市場銷量,動力電池用鋰離子電池正極材料的市場需求也隨之增加。目前商業化最成熟的正極材料是鈷酸鋰(licoo2),它具有較好的電化學性能,但安全性不好且價格昂貴,不適用于動力電池;尖晶石錳酸鋰(limn2o4)、磷酸鐵鋰(lifepo4)、二元材料(linixco1-xo2)及三元材料(linixcoymn1-x-yo2)等已在動力電池中得到應用。磷酸鐵鋰安全性高、循環壽命長,但產品存在一致性不好,極片加工性能及低溫性能較差等問題;二元材料、三元材料克容量高,但價格相對較高、安全性差;錳酸鋰具有原材料錳儲量豐富、成本低、倍率性能好、安全性好的優點,但循環性能較差,特別是高溫循環性能差,制約了其應用;錳酸鋰循環性能不好的主要原因在于,隨著充放電過程中鋰的脫出與插入,錳酸鋰晶體結構不斷發生變化,導致晶格穩定性降低及錳在電解液中溶解。
當前錳酸鋰大多采用電解二氧化錳為錳源,通過與碳酸鋰或氫氧化鋰、添加劑混合,在高溫下焙燒而制得,得到的錳酸鋰性能不夠好,主要缺點是電解二氧化錳粉末由機械粉碎制得,其顆粒形貌不好控制,通常為不定型顆粒,比表面積大;另外電解二氧化錳雜質含量高,如硫酸根一般為1.2%以上,鈣雜質在300ppm左右,鈉雜質含量在3000ppm的水平,且在粉碎過程中因機械設備問題容易帶入鐵雜質,這些因素都對錳酸鋰性能造成負面影響。
為提高錳酸鋰結構穩定性,可采用摻雜改性方法來改進,通常是將鋰源、錳源、摻雜元素的化合物混合后經高溫焙燒而制得,但這有可能導致摻雜元素分布不均勻,主要集中在顆粒表面,顆粒內部的摻雜效果得不到保障。
目前最需要改進錳酸鋰材料的循環性能,特別是高溫循環性能,由于原材料純度及摻雜改性工藝等方面的原因,尚不能從根本上解決錳酸鋰循環性能較差的問題。
技術實現要素:
本發明要解決的技術問題是:提供一種雜質含量低、摻雜元素分子級均勻分布的球形摻雜錳酸鋰的制備方法,該方法能夠解決錳酸鋰循環性能較差的問題。
解決上述技術問題的技術方案是:一種球形摻雜錳酸鋰的制備方法,球形摻雜錳酸鋰的化學式為li1+xmn2-ymy-xo4,其中m是摻雜元素,0≤x≤0.1,0≤y-x≤0.5,
該球形摻雜錳酸鋰的制備方法包括以下步驟:
(1)稱取摻雜元素可溶性鹽和二價錳的可溶性錳鹽用水配制成含摻雜元素離子和二價錳離子的混合溶液;混合溶液中,二價錳離子的濃度為20g/l~200g/l,摻雜元素離子的濃度為0.05g/l~50g/l,
(2)稱取氫氧化鈉,用水配制成濃度為50g/l~500g/l的氫氧化鈉溶液;
(3)將上述步驟(1)中的混合溶液和步驟(2)中的氫氧化鈉溶液連續地加入到反應器中,控制反應器中二價錳離子與摻雜元素離子的總摩爾數與氫氧化鈉的摩爾數比為(0.5~1.5)∶2,進行共沉淀反應,控制反應溫度為40℃~80℃,反應液的ph值為7.5~13.5,同時向反應液中加入足量的氧化劑對反應過程中生成的沉淀物進行氧化處理,然后用水漂洗分離后的沉淀物,再經干燥得到球形摻雜四氧化三錳;所述的氧化劑是雙氧水、氧氣、空氣、過硫酸銨或硫代硫酸鈉中的至少一種;
(4)按化學式中的化學計量比稱取碳酸鋰或氫氧化鋰與步驟(3)制得的球形摻雜四氧化三錳充分混合,然后在氧氣或空氣氣氛中經700℃~1000℃高溫焙燒5~30小時,得到球形摻雜錳酸鋰;
在步驟(1)制得的混合溶液中添加調和劑a或是在步驟(2)制得的氫氧化鈉溶液中添加調和劑b或是既在步驟(1)制得的混合溶液中添加調和劑a,又在步驟(2)制得的氫氧化鈉溶液中添加調和劑b;所述調和劑a是司本-80、op-10、十二烷基硫酸鈉、氟化銨、氯化銨、硫酸銨、硝酸銨、氟化鈉、乙醇、乙二醇、異丙醇、正丁醇、乙二胺四乙酸、乙二胺四乙酸二鈉、乙二胺四乙酸四鈉、羧甲基纖維素、羧甲基纖維素鈉和聚乙烯醇中的至少一種;所述調和劑b包括司本-80、op-10、十二烷基硫酸鈉、氨水、乙二胺、氟化銨、氯化銨、硫酸銨、硝酸銨、氟化鈉、乙醇、乙二醇、異丙醇、正丁醇、edta、edta二鈉、edta四鈉、羧甲基纖維素、羧甲基纖維素鈉和聚乙烯醇中的至少一種;加入調和劑a后,混合溶液中調和劑a的濃度控制在0.1g/l~20g/l,加入調和劑b后,氫氧化鈉溶液中調和劑b的濃度控制在0.1g/l~50g/l。
化學式中m是li、ni、mg、al、co、cr、ti元素中的至少一種。
步驟(4)中的焙燒溫度為800~900℃,焙燒時間為10~30小時。
步驟(1)中所述的可溶性錳鹽為硫酸錳、硝酸錳和氯化錳中的至少一種。
步驟(1)中所述的摻雜元素可溶性鹽為硫酸鹽、硝酸鹽和氯化鹽中的至少一種。
本發明的進一步技術方案是:向反應液中加入足量的氧化劑對反應過程中生成的沉淀物進行氧化處理的具體操作步驟為:向反應液中加入質量濃度為5~15%氧化劑進行氧化處理5~20小時,氧化劑的用量以控制反應液的ph值為7.5~13.5為準,所述的氧化劑是雙氧水、過硫酸銨溶液和硫代硫酸鈉溶液中的至少一種。
與現有技術相比,本發明的優點在于:本發明制備方法的工藝流程簡單,易于實現規模生產;在本發明中,首先通過濕法工藝制備雜質含量低,摻雜元素分子級均勻分布的球形摻雜四氧化三錳前驅體,然后以球形摻雜四氧化三錳為錳源制備出球形摻雜錳酸鋰。可通過控制原材料的雜質水平而降低產品的雜質含量,制造出微觀結構為球形的粉末顆粒,實現了分子水平的均勻摻雜效果。使用本發明方法制造出的球形摻雜錳酸鋰產品,具有立方尖晶石結構,空間群fd-3m(no.227)的錳酸鋰顆粒;其雜質含量低,鉀、鈉、鈣雜質含量均小于50ppm,鐵、銅雜質含量小于20ppm,硫酸根含量小于0.1%;摻雜元素在錳酸鋰顆粒內部為分子級均勻分布,能夠提高錳酸鋰結構穩定性;其微觀二次顆粒形貌為球形,粒度d50為5μm~20μm;比表面積小于0.8m2/g,振實密度大于2.0g/cm3;經55℃高溫循環200次,容量保持率大于95%,可顯著改進錳酸鋰的循環性能,特別是高溫循環性能,而且可增強錳酸鋰在容量型鋰離子電池和動力電池中的應用,是高端鋰離子電池的理想正極材料。
與將鋰源、錳源(氧化錳、氫氧化錳或碳酸錳)和摻雜化合物固相混合后高溫焙燒制備錳酸鋰的傳統工藝相比,本發明雜質含量更低,摻雜元素在顆粒內部分布更均勻;與濕法制備氧化錳、氫氧化錳或碳酸錳前驅體,再將錳前驅體、鋰源和摻雜化合物混合后高溫焙燒制備錳酸鋰的工藝相比,本發明摻雜元素在顆粒內部分布更均勻;與濕法制備氧化錳、氫氧化錳或碳酸錳,在其表面沉淀摻雜元素化合物得到摻雜錳前驅體,再將鋰源和摻雜錳前驅體混合后高溫焙燒制備錳酸鋰的工藝相比,本發明摻雜元素在顆粒內部分布更均勻。
附圖說明
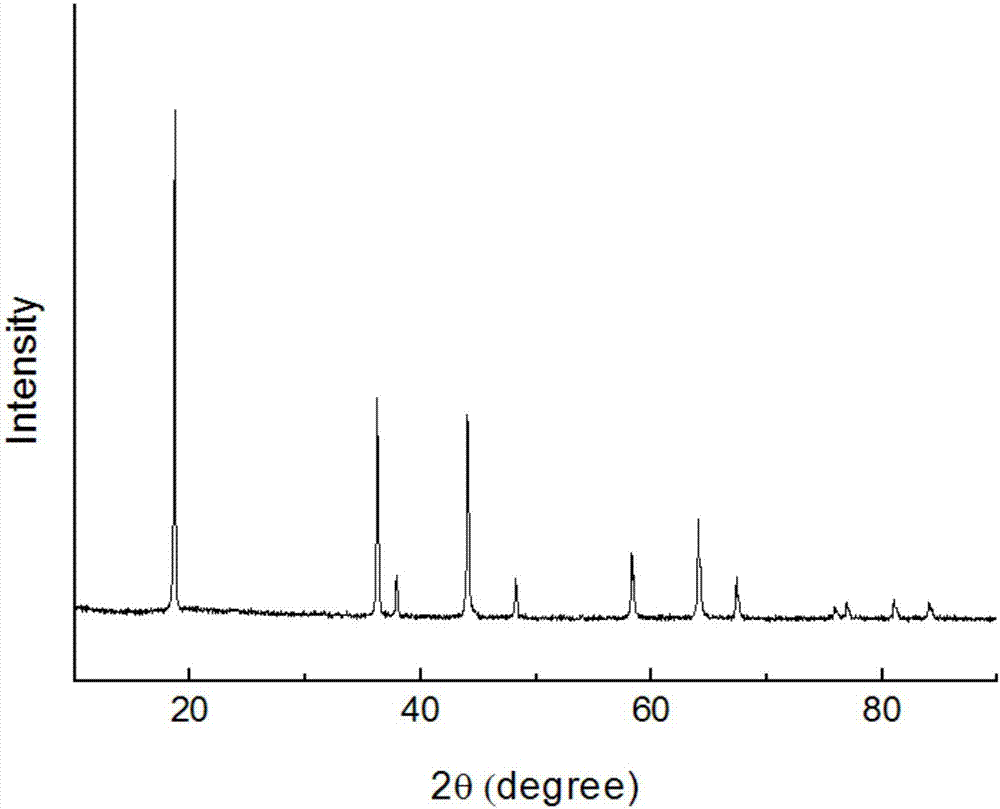
圖1為本發明實施例1中制得的球形摻雜錳酸鋰的xrd衍射圖。
圖2為本發明實施例1中制得的球形摻雜錳酸鋰的sem圖。
圖3為本發明實施例4中制得的球形摻雜錳酸鋰的sem圖。
圖4為本發明實施例1中制得的球形摻雜錳酸鋰的循環曲線圖。
具體實施方式
一種球形摻雜錳酸鋰的制備方法,球形摻雜錳酸鋰的化學式為li1+xmn2-ymy-xo4,其中m是摻雜元素,m是li、ni、mg、al、co、cr、ti等元素中的至少一種,0≤x≤0.1,0≤y-x≤0.5。球形摻雜錳酸鋰是具有立方尖晶石結構,空間群fd-3m(no.227)的錳酸鋰顆粒;其雜質含量低,鉀、鈉、鈣雜質含量均小于50ppm,鐵、銅雜質含量小于20ppm,硫酸根含量小于0.1%;摻雜元素在錳酸鋰顆粒內部為分子級均勻分布,能夠提高結構穩定性;其微觀二次顆粒形貌為球形,粒度d50為5μm~20μm;比表面積小于0.8m2/g,振實密度大于2.0g/cm3。
該球形摻雜錳酸鋰的制備方法包括以下步驟:
(1)按化學式中的化學計量比稱取摻雜元素可溶性鹽和二價錳的可溶性錳鹽用純水或去離子水配制成含摻雜元素離子和二價錳離子的混合溶液;混合溶液中,二價錳離子的濃度為20g/l~200g/l,摻雜元素離子的濃度為0.05g/l~50g/l,可溶性錳鹽為硫酸錳、硝酸錳、氯化錳中的至少一種,例如可以是前述可溶性錳鹽中的一種,也可以是其中任意二種錳鹽的任意比例的混合,還可以是其中三種的任意比例的混合。摻雜元素可溶性鹽為硫酸鹽、硝酸鹽、氯化鹽中的至少一種。例如可以是前述摻雜元素可溶性鹽中的一種,也可以是其中任意兩種的任意比例的混合,還可以是其中三種的任意比例的混合。
(2)稱取調和劑a,將其加入到步驟(1)得到的混合溶液中,攪拌均勻后形成含調和劑a濃度為0.1g/l~20g/l的混合溶液;調和劑a為司本-80、op-10、十二烷基硫酸鈉、氟化銨、氯化銨、硫酸銨、硝酸銨、氟化鈉、乙醇、乙二醇、異丙醇、正丁醇、edta、edta二鈉、edta四鈉、羧甲基纖維素、羧甲基纖維素鈉和聚乙烯醇中的至少一種(根據調和劑b的使用情況,調和劑a可以不加);配制的錳鹽溶液中,如果有沉淀或雜質,需要先進行過濾或除雜處理,去除雜質的最好時機是在配制前對原材料進行除雜處理。
(3)稱取氫氧化鈉,用純水或去離子水配制成濃度為50g/l~500g/l的氫氧化鈉溶液(所用氫氧化鈉可以是市售的固體片堿,也可以直接用市售的液堿配制)。
(4)稱取調和劑b,將其加入到步驟(3)得到的氫氧化鈉溶液中,攪拌均勻后形成含調和劑b濃度為0.1g/l~50g/l的氫氧化鈉溶液;調和劑b為司本-80、op-10、十二烷基硫酸鈉、氨水、乙二胺、氟化銨、氯化銨、硫酸銨、硝酸銨、氟化鈉、乙醇、乙二醇、異丙醇、正丁醇、edta、edta二鈉、edta四鈉、羧甲基纖維素、羧甲基纖維素鈉和聚乙烯醇中的至少一種(可視情況不加調和劑b);配制的氫氧化鈉溶液或加入調和劑b后的氫氧化鈉溶液,如果有沉淀或雜質,需要在配制后進行過濾或除雜處理,為使最終產品的雜質含量最低,最好是在配制前對原材料進行除雜處理。
(5)將上述步驟(2)中的混合溶液和步驟(4)中的氫氧化鈉溶液連續地加入到反應器中(即一邊反應的同時一邊按比例加入),控制反應器中二價錳離子與摻雜元素離子的總摩爾數與氫氧化鈉的摩爾數比為(0.5~1.5)∶2,進行共沉淀反應,控制反應溫度為40℃~80℃,反應液的ph值為7.5~13.5,同時向反應液中加入足量的氧化劑對反應過程中生成的沉淀物進行氧化處理,使二價錳化合物沉淀轉化為四氧化三錳;反應過程為連續加料時,反應產物自反應器上的溢流口排出;然后用純水或去離子水漂洗分離后的沉淀物,再經干燥得到球形摻雜四氧化三錳;所述的氧化劑是雙氧水、氧氣、空氣、過硫酸銨或硫代硫酸鈉中的至少一種;氧化劑的用量不能低于反應的理論需要量。
(6)按化學式中的化學計量比稱取碳酸鋰或氫氧化鋰與步驟(5)制得的球形摻雜四氧化三錳充分混合,然后在氧氣或空氣氣氛中經700℃~1000℃高溫焙燒5~30小時,得到球形摻雜錳酸鋰。
上述的調和劑a和/或調和劑b的作用都是便于后續步驟(5)中的沉淀生長為球形,機理上可以是通過選擇性吸附來改變沉淀顆粒的生長方向,也可以是通過改變沉淀顆粒與反應液的界面潤濕性能來改善晶粒的生長速度,還可以是影響反應速度來控制沉淀顆粒的生長速度;無論對于調和劑a和/或調和劑b,如果選用物質的溶解速度慢,則均可進行加熱來加速溶解。
為檢測本發明的球形摻雜錳酸鋰的物化性能,用法國jy公司ultima電感耦合等離子發射光譜儀(icp-aes)對材料雜質含量進行定量分析;用荷蘭x’pertpro-mpd型xrd衍射儀進行結構測試;用日本jeol公司jsm-6360lv型掃描電鏡sem進行顆粒形貌與大小分析;用英國malvern儀器有限公司mastersizer2000型激光粒度分析儀進行粒度分析;用美國quantachrome公司的nova-1000型比表面測試儀進行比表面積分析。
為檢測本發明的球形摻雜錳酸鋰的電化學性能,將其組裝成cr2032型扣式電池進行電化學性能測試,用本發明的錳酸鋰材料90%(重量百分數),導電碳黑5%,粘結劑pvdf(聚偏氟乙烯)5%,混合調成漿狀,涂在鋁箔上,在120℃干燥,制成電極;以金屬鋰片為負極,電解液為1mol/llipf6/ec+dmc(體積比1:1),ec為碳酸乙烯酯,dmc為碳酸二甲酯,組裝cr2032型扣式電池。用武漢藍電電池測試系統進行充放電測試,測試溫度55℃,充放電電壓范圍為3.0~4.3v,首次充放電測試倍率為0.2c,第2次~第201次循環為0.5c恒流恒壓充電,1c恒流放電。
下面結合具體實施例對本發明作進一步的描述。
實施例1:
一種本發明的球形摻雜錳酸鋰,其化學式為li1.05mn1.80al0.15o4。
用純水配制二價錳離子濃度為60g/l與三價鋁離子濃度為2.5g/l的硫酸錳與硫酸鋁混合溶液20l,稱取60g異丙醇和70g氟化鈉加入到上述溶液中并攪拌均勻;用純水配制濃度為200g/l的氫氧化鈉溶液20l,按硫酸錳與硫酸鋁混合溶液500ml/h的流量、錳離子與鋁離子的摩爾數之和與氫氧化鈉的摩爾數之比為1∶2的比例,用計量泵將混合溶液和氫氧化鈉溶液加入至容積為10l的反應器中,控制反應的溫度為70℃,反應液的ph值為10.0,同時通足量的空氣對沉淀進行氧化,反應進行30小時后停止,水洗、干燥后,得到摻鋁四氧化三錳前驅體。所得的摻鋁四氧化三錳粒度d50為17.36μm,振實密度為2.6g/cm3。
將上述球形摻鋁四氧化三錳、碳酸鋰以mn:li摩爾比=1.80:1.05的比例稱量,充分混合后在空氣氣氛中經820℃高溫焙燒15小時,得到錳酸鋰產品。
所得錳酸鋰的雜質檢測結果為鉀6ppm、鈉38ppm、鈣42ppm、鐵8ppm、銅3ppm,硫酸根0.08%;圖1為xrd衍射圖,所得錳酸鋰沒有雜相;圖2為sem圖,所得錳酸鋰二次顆粒形貌為球形;其粒度d50為18.58μm,比表面積為0.41m2/g,振實密度為2.1g/cm3。
以所得的錳酸鋰為正極活性物質,制作扣式電池,進行55℃高溫測試。圖4為其循環曲線,如圖所示,首次0.2c放電克容量為106.6mah/g,第2次循環1c放電克容量為105.9mah/g,1c循環200次后容量保持率為95.4%。
實施例2:
一種本發明的球形摻雜錳酸鋰,其化學式為li1.05mn1.85al0.10o4。
用純水配制二價錳離子濃度為60g/l與三價鋁離子濃度為1.6g/l的硫酸錳與硫酸鋁混合溶液20l,稱取60g異丙醇和70g氟化鈉加入到上述溶液中并攪拌均勻;用純水配制濃度為200g/l的氫氧化鈉溶液20l,按硫酸錳與硫酸鋁混合溶液500ml/h的流量、錳離子與鋁離子的摩爾數之和與氫氧化鈉的摩爾數之比為1∶2的比例,用計量泵將混合溶液和氫氧化鈉溶液加入至容積為10l的反應器中,反應液的ph值為10.0,控制反應的溫度為70℃,同時加入質量濃度為10%的雙氧水對沉淀進行氧化,雙氧水的用量以控制氧化過程反應液的ph值為9.5~10.5為宜,反應進行5小時后停止,水洗、干燥后,得到摻鋁四氧化三錳前驅體。所得的摻鋁四氧化三錳粒度d50為17.15μm,振實密度為2.6g/cm3。
將上述球形摻鋁四氧化三錳、碳酸鋰以mn:li摩爾比=1.85:1.05的比例稱量,充分混合后在空氣氣氛中經820℃高溫焙燒15小時,得到錳酸鋰產品。
所得錳酸鋰的雜質檢測結果為鉀7ppm、鈉42ppm、鈣40ppm、鐵9ppm、銅2ppm,硫酸根0.06%;粒度d50為18.03μm,比表面積為0.45m2/g,振實密度為2.1g/cm3。55℃高溫測試,首次0.2c放電克容量為113.1mah/g,第2次循環1c放電克容量為112.7mah/g,1c高溫循環200次后容量保持率為95.1%。
實施例3:
一種本發明的球形摻雜錳酸鋰,其化學式為li1.06mn1.84co0.05al0.05o4。
用純水配制二價錳離子濃度為75g/l、二價鈷離子濃度為2.2g/l與三價鋁離子濃度為1.0g/l的硫酸錳、硫酸鈷與硫酸鋁混合溶液20l,稱取80g無水乙醇加入到上述溶液中并攪拌均勻;用純水配制濃度為120g/l的氫氧化鈉溶液20l,稱取質量分數為25%的氨水300g加入到氫氧化鈉溶液中并攪拌均勻,按硫酸錳、硫酸鈷與硫酸鋁混合溶液800ml/h的流量,錳離子、鈷離子與鋁離子的摩爾數之和與氫氧化鈉的摩爾數之比為1∶2的比例,用計量泵將混合溶液和氫氧化鈉溶液加入至容積為10l的反應器中,控制反應的溫度為65℃,反應液的ph值為9.5,同時加入質量濃度為10%的硫代硫酸鈉溶液對沉淀進行氧化,硫代硫酸鈉溶液的用量以控制氧化過程反應液的ph值為9~10為宜,反應進行8小時后停止,水洗、干燥后,得到摻鈷鋁四氧化三錳前驅體。所得的摻鈷鋁四氧化三錳粒度d50為7.17μm,振實密度為2.3g/cm3。
將上述球形摻鈷鋁四氧化三錳、氫氧化鋰以mn:li摩爾比=1.84:1.06的比例稱量,充分混合后在氧氣氣氛中經850℃高溫焙燒15小時,得到錳酸鋰產品。
所得錳酸鋰的雜質檢測結果為鉀2ppm、鈉46ppm、鈣42ppm、鐵10ppm、銅2pm,硫酸根0.07%;粒度d50為8.71μm,比表面積為0.56m2/g,振實密度為2.6g/cm3;55℃高溫測試,首次0.2c放電克容量為110.5mah/g,第2次循環1c放電克容量為110.0mah/g,1c高溫循環200次后容量保持率為95.3%。
實施例4:
一種本發明的球形摻雜錳酸鋰,其化學式為li1.03mn1.89co0.05ni0.03o4。
用純水配制二價錳離子濃度為60g/l、二價鈷離子濃度為1.7g/l與二價鎳離子濃度為1.0g/l的氯化錳、氯化鈷與氯化鎳混合溶液30l;用純水配制濃度為180g/l的氫氧化鈉溶液30l,稱取120g氯化銨與80g氟化鈉加入到氫氧化鈉溶液中并攪拌溶解完全,按氯化錳、氯化鈷與氯化鎳混合溶液700ml/h的流量,錳離子、鈷離子與鎳離子的摩爾數之和與氫氧化鈉的摩爾數之比為1∶2的比例,用計量泵將混合溶液和氫氧化鈉溶液加入至容積為10l的反應器中,控制反應的溫度為55℃,反應液的ph值為9.2,同時加入質量濃度為10%的過硫酸銨溶液對沉淀進行氧化,過硫酸銨溶液的用量以控制氧化過程反應液的ph值為9~10為宜,反應進行10小時后停止,水洗、干燥后,得到摻鈷鎳四氧化三錳前驅體。所得的摻鈷鎳四氧化三錳粒度d50為9.08μm,振實密度為2.4g/cm3。
將上述球形摻鈷鎳四氧化三錳、氫氧化鋰以mn:li摩爾比=1.89:1.03的比例稱量,充分混合后在空氣氣氛中經810℃高溫焙燒20小時,得到錳酸鋰產品。
所得錳酸鋰的雜質檢測結果為鉀3ppm、鈉36ppm、鈣37ppm、鐵17ppm、銅6pm,硫酸根0.05%;圖3為sem圖,所得錳酸鋰二次顆粒形貌為球形,粒度d50為11.21μm;比表面積為0.51m2/g,振實密度為2.0g/cm3;55℃高溫測試,首次0.2c放電克容量為116.1mah/g,第2次循環1c放電克容量為115.5mah/g,1c高溫循環200次后容量保持率為95.4%。
- 還沒有人留言評論。精彩留言會獲得點贊!