一種表面改性的鋰電池高鎳正極材料的制備方法及產品與流程
本發明屬于二次鋰離子電池領域,更具體地,涉及一種表面改性的鋰電池高鎳正極材料的制備方法及產品。
背景技術:
隨著社會的不斷進步,在全球經濟飛速增長的現在,科學技術的不斷發展帶來的產品在人們的生活中起著不可或缺的作用,電子產品,電動汽車,醫療設備等對儲能的要求越來越高,人們對能源的需求日益增加,對社會與經濟可持續發展的重要性的認識不斷深化。鋰離子電池作為新一代的綠色高能充電電池,自1990年問世以來,以其電壓高、能量密度大、循環性能好、自放電小和環境友好等突出優點,近20年取得了迅猛發展,已被廣泛用作袖珍貴重家用電器如移動電話、便攜式計算機、攝像機、照相機等的電源,目前鋰離子電池正極材料主要包括鈷酸鋰、磷酸鐵鋰、高鎳正極材料等。
然而,進一步的研究表明,鋰離子電池中,由于正極材料所處的電勢較高,且脫鋰態正極材料具有較強的氧化性,易與有機電解液發生副反應,以至于惡化電池的性能。雖然鎳系正極材料linio2由于具有放電比容量高、價格低廉等優點,然而其固有的一些缺陷也限制了其廣泛應用:如計量比的linio2難以合成、放電過程中存在較多雜相變、ni2+占據li+的3a位置導致陽離子混排、高鎳含量在充電末期催化活性很強,易催化電解液分解、室溫及儲存性能較差,此外,高鎳正極材料在充放電過程中存在首圈效率不高以及循環過程中由于結構由穩定的層狀結構轉變成不穩定的巖鹽相,導致循環性能差的問題。
由于存在上述缺陷和不足,本領域亟需對高鎳正極材料作出進一步的完善和改進,解決其在充放電過程中存在的循環性能差的問題。
技術實現要素:
針對現有技術的以上缺陷或改進需求,本發明提供了一種表面改性的鋰電池高鎳正極材料的制備方法及該材料,該制備方法結合高鎳正極材料自身的性能特點,采用原子層沉積技術對高鎳正極材料進行表面包覆改性,得到表面改性的鋰離子高鎳正極材料,進而采用該材料制造鋰離子二次電池,由此制得的鋰離子二次電池可克服高鎳正極材料在充放電過程中容量衰減的問題,其結構穩定性和循環性能大大提高,此外本申請的表面改性的鋰電池高鎳正極材料還具有制備方法簡單易行、包覆層厚度易控制、適合大規模生產等優點。
為實現上述目的,按照本發明的一個方面,提出了一種表面改性的鋰電池高鎳正極材料的制備方法,包括以下步驟:
(a)將高鎳正極材料粉末放入原子層沉積系統的反應腔體中,并將所述反應腔體抽真空5s~10s,所述高鎳正極材料為linixcoymzo2,其中0.6≤x≤1,0≤y≤0.4,0≤z≤0.4,且x+y+z=1,m為mn、al、mg、ti中的一種或幾種;升高所述反應腔體的溫度,使得溫度保持在140℃~160℃;
(b)向所述反應腔體中通入反應源,并使得反應腔體中的壓力達到5mbar~8mbar,該反應源吸附在所述高鎳正極材料粉末的表面;
(c)向所述反應腔體中通入n2,以帶走所述反應腔體中過剩的反應源,通入時間為120s~150s;
(d)繼續向所述反應腔體中通入h2o,直至所述反應腔體的壓力達到5mbar~8mbar時停止通入,該h2o與吸附在所述高鎳正極材料表面的所述反應源發生反應,以在所述高鎳正極材料的表面沉積獲得氧化物薄膜;
(e)然后向所述反應腔體中通入n2,以帶走所述反應腔體中的反應副產物和過剩的h2o,通入時間為120s~150s;
(f)重復步驟(b)~(e)獲得具有所需沉積厚度的表面改性的鋰電池高鎳正極材料。
作為進一步優選的,所述反應源為金屬有機物、金屬單質和金屬鹵化物中的一種;所述金屬有機物、金屬單質和金屬鹵化物中采用的金屬元素為ti、al、fe、zn中的一種。
按照本發明的另一方面,提供了一種表面改性的鋰電池高鎳正極材料,其由所述的方法制備。
按照本發明的另一方面,提供了一種正極極片,所述正極極片包括集流體和涂覆在該集流體上的所述的表面改性的鋰電池高鎳正極材料。
作為進一步優選的,所述正極極片還包括導電劑和粘結劑,所述導電劑、粘結劑和表面改性的鋰電池高鎳正極材料三者混合后涂覆在所述集流體上。
作為進一步優選的,所述表面改性的鋰電池高鎳正極材料、導電劑、粘結劑的混合比例為:表面改性的鋰電池高鎳正極材料的質量分數為50~99.5wt%,導電劑為0.1~40wt%,粘結劑為0.1~40wt%。
作為進一步優選的,所述導電劑為炭黑、乙炔黑、天然石墨、碳納米管、石墨烯、碳纖維中的一種或多種;所述粘結劑為聚四氟乙烯、聚偏二氟乙烯、聚氨酯、聚丙烯酸、聚酰胺、聚丙烯、聚乙烯基醚、聚酰亞胺、苯乙烯-丁二烯共聚物、羧甲基纖維素鈉中的一種或多種。
按照本發明的另一方面,提供了一種鋰離子二次電池,其包括所述的正極極片。
作為進一步優選的,還包括負極極片、隔膜、電解液和電池殼。
作為進一步優選的,所述隔膜優選為芳綸隔膜、無紡布隔膜、聚乙烯微孔膜、聚丙烯膜、聚丙烯聚乙烯雙層或三層復合膜、陶瓷涂覆層隔膜中的一種;所述電解液包括電解質和溶劑,所述電解質優選為lipf6、libf4、liclo4、liasf6、licf3so3、lin(cf3so2)、libob、licl、libr、lii中的一種或者多種;所述溶劑優選為丙烯碳酸酯(pc)、碳酸二甲酯(dmc)、碳酸甲乙酯(emc)、1,2-二甲氧基乙烷(dme)、碳酸乙烯酯、碳酸丙烯酯、碳酸丁烯酯、碳酸二乙酯、碳酸甲丙酯、乙腈、乙酸乙酯、亞硫酸乙烯酯中的一種或多種。
總體而言,通過本發明所構思的以上技術方案與現有技術相比,主要具備以下的技術優點:
1.本發明所述的制備方法,利用ald對用于制備電池正極的高鎳正極材料進行表面改性,方法簡單易操作,能夠進行大規模的包覆,且與現有方法相比,包覆層的均勻程度更好,包覆層的厚度更好控制。
2.本發明在表面改性的過程中,將反應溫度、反應時間和反應壓力等關鍵參數控制在一定范圍內,能夠有效地提高表面改性的效果,同時提高表面改性的反應速度,從而最終提高由此高鎳正極材料制備的鋰離子二次電池的性能。
3.本發明所述的制備方法,選用了過渡金屬氧化物例如tio2來對高鎳正極材料進行表面處理,通過簡單的幾個步驟對高鎳正極材料進行了表面改性處理,簡化了整個制備方法的流程,高效率、高質量地獲得了所要制備的正極材料,并且整個制備過程便于操作和質量控制,尤其是對于表面包覆的控制尤為的簡單易行。
附圖說明
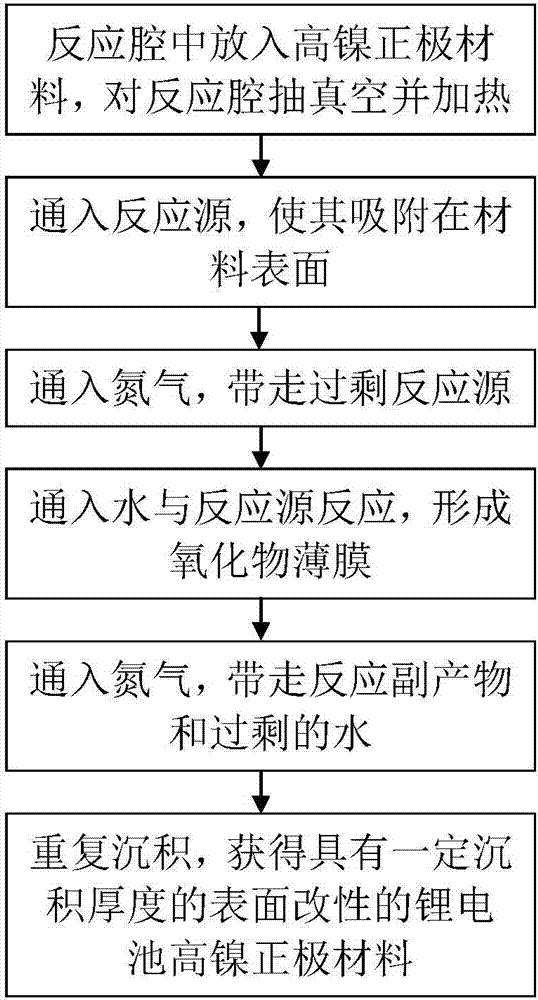
圖1是本發明的一種表面改性的鋰電池高鎳正極材料的制備方法的流程圖;
圖2是未包覆的高鎳正極材料和包覆不同厚度的高鎳正極材料的x射線粉末衍射(xrd)圖譜;
圖3(a)和圖3(b)是未包覆的高鎳正極材料的掃描電子顯微鏡(sem)圖;
圖3(c)和圖3(d)是本發明包覆的高鎳正極材料的掃描電子顯微鏡(sem)圖;
圖4(a)是未包覆的高鎳正極材料的tem圖;圖4(b)是本發明包覆的高鎳正極材料的透射電子顯微鏡(tem)圖;
圖5(a)是未包覆的高鎳正極材料和包覆不同厚度的高鎳正極材料的電池的首圈充放電曲線;
圖5(b)是未包覆的高鎳正極材料和包覆不同厚度的高鎳正極材料的電池的循環性能圖;
圖5(c)是未包覆的高鎳正極材料和包覆的高鎳正極材料電池在不同電流密度下的倍率性能;
圖5(d)是未包覆的高鎳正極材料和包覆的高鎳正極材料電池在不同倍率下的放電曲線。
具體實施方式
為了使本發明的目的、技術方案及優點更加清楚明白,以下結合附圖及實施例,對本發明進行進一步詳細說明。應當理解,此處所描述的具體實施例僅僅用以解釋本發明,并不用于限定本發明。此外,下面所描述的本發明各個實施方式中所涉及到的技術特征只要彼此之間未構成沖突就可以相互組合。
本發明的原理是利用原子層沉積法在正極材料表面沉積金屬氧化物薄膜,通過這種改性方法能夠獲得較為均勻的包覆層,并且能夠有效的控制包覆層的厚度,通過包覆表面薄膜能夠有效的隔絕電解液多材料表面的侵蝕,提高了整體的材料的結構穩定性。
如圖1所示,本發明提供了一種表面改性的鋰電池高鎳正極材料的制備方法,包括以下步驟:
(a)將高鎳正極材料放入ald(atomiclayerdeposition,原子層沉積)系統的反應腔體中,并將反應腔體抽真空5s~10s,高鎳正極材料為linixcoymzo2,其中0.6≤x≤1,0≤y≤0.4,0≤z≤0.4,且x+y+z=1,m為mn、al、mg、ti中的一種或幾種,升高反應腔體的溫度,使得反應溫度始終保持在140℃~160℃;
(b)向抽真空后的反應腔體中通入反應源,并使得反應腔體中的壓力達到5mbar~8mbar,該反應源吸附在所述高鎳正極材料的表面;
(c)向所述反應腔體中通入n2,以帶走所述反應腔體中過剩的反應源,通入時間為120s~150s;
(d)繼續向所述反應腔體中通入h2o,當腔體壓力達到5mbar~8mbar時結束通入h2o,由于反應腔體中的溫度為140℃~160℃,通入的水在該溫度下變為水蒸氣,水蒸氣與吸附在高鎳正極材料表面的反應源發生反應,以在高鎳正極材料的表面沉積一氧化物薄膜,反應過程中會產生反應副產物,具體的副產物在此不贅述;
(e)然后向反應腔體中通入n2,以帶走所述反應腔體中的反應副產物和過剩的h2o,通入時間為120s~150s;
(f)重復步驟(b)~(e)獲得具有所需沉積厚度的表面改性的鋰電池高鎳正極材料。
具體的,沉積的反應源為金屬有機物、金屬單質和金屬鹵化物中的一種,其中,金屬元素為ti、al、fe、zn中的一種。上述反應源具有以下幾個特點:1)反應源有足夠高的蒸氣壓,能夠充分覆蓋和填充基體材料的表面;2)反應源有較好的穩定性,不會發生自分解,也不會腐蝕基體材料;3)反應源有一定的化學活性,能夠迅速的在材料表面形成化學吸附,能夠在短時間內達到飽和吸附。
本發明還提供了一種正極極片,該正極極片包括集流體和涂覆在該集流體上的由本發明方法制備的所述鋰電池高鎳正極材料。
進一步的,該正極極片還包括導電劑和粘結劑,所述導電劑、粘結劑和鋰電池高鎳正極材料三者混合后涂覆在所述集流體上。
優選的,鋰電池高鎳正極材料、導電劑、粘結劑的混合比例為:鋰電池高鎳正極材料的質量分數為50~99.5wt%,導電劑為0.1~40wt%,粘結劑為0.1~40wt%,較多的比較試驗表明,采用該質量比制備的正極極片具有較為優良的電化學性能。
進一步優選的,所述導電劑為炭黑、乙炔黑、天然石墨、碳納米管、石墨烯、碳纖維中的一種或多種;所述粘結劑為聚四氟乙烯、聚偏二氟乙烯、聚氨酯、聚丙烯酸、聚酰胺、聚丙烯、聚乙烯基醚、聚酰亞胺、苯乙烯-丁二烯共聚物、羧甲基纖維素鈉中的一種或多種。
本發明還提供了一種鋰離子二次電池,其包括由本發明方法制備的所述正極極片。
具體的,該鋰離子二次電池還包括負極極片、隔膜、電解液和電池殼。
優選的,所述隔膜為芳綸隔膜、無紡布隔膜、聚乙烯微孔膜、聚丙烯膜、聚丙烯聚乙烯雙層或三層復合膜、陶瓷涂覆層隔膜中的一種。
進一步優選的,電解液包括電解質和溶劑,電解質優選為lipf6、libf4、liclo4、liasf6、licf3so3、lin(cf3so2)、libob、licl、libr、lii中的一種或者多種;溶劑優選為丙烯碳酸酯(pc)、碳酸二甲酯(dmc)、碳酸甲乙酯(emc)、1,2-二甲氧基乙烷(dme)、碳酸乙烯酯、碳酸丙烯酯、碳酸丁烯酯、碳酸二乙酯、碳酸甲丙酯、乙腈、乙酸乙酯、亞硫酸乙烯酯中的一種或多種。
以下為本發明的具體實施例。
實施例1
利用ald設備在高鎳正極材料linio2表面沉積tio2薄膜,反應源為h2o和異丙基鈦,沉積溫度為150℃。將高鎳正極材料linio2放入到ald中的反應腔中,沉積過程如下:(1)將反應腔抽真空5s;(2)通入反應鈦源(異丙基鈦)使得腔體壓力達到5mbar;(3)向反應體系中通入n2,時間為120s,用來帶走反應的副產物和過剩的異丙基鈦;(4)繼續向反應腔內通入h2o,當腔體壓力達到5mbar時結束;(5)向反應體系中通入n2,時間為120s,用來帶走反應的副產物和過剩的h2o;步驟(1)~(5)為一個完整沉積過程,循環次數為20次。
實施例2
利用ald設備在高鎳正極材料lini0.6co0.4o2表面沉積zno薄膜,反應源為h2o和zncl2,沉積溫度為150℃。將高鎳正極材料lini0.6co0.4o2放入到ald中的反應腔中,沉積過程如下:(1)將反應腔抽真空6s;(2)通入反應鋅源(zncl2)使得腔體壓力達到6mbar;(3)向反應體系中通入n2,時間為130s,用來帶走反應的副產物和過剩的氯化鋅;(4)繼續向反應腔內通入h2o,當腔體壓力達到6mbar時結束;(5)向反應體系中通入n2,時間為140s,用來帶走反應的副產物和過剩的h2o;步驟(1)~(5)為一個完整沉積過程。,循環次數為25次。
實施例3
利用ald設備在高鎳正極材料lini0.4co0.2mn0.4o2表面沉積al2o3薄膜,反應源為h2o和金屬鋁,沉積溫度為150℃。將高鎳正極材料lini0.4co0.2m0.4o2放入到ald中的反應腔中,沉積過程如下:(1)將反應腔抽真空10s;(2)通入反應鋁源(鋁單質)使得腔體壓力達到8mbar;(3)向反應體系中通入n2,時間為150s,用來帶走反應的副產物和過剩的金屬鋁;(4)繼續向反應腔內通入h2o,當腔體壓力達到8mbar時結束;(5)向反應體系中通入n2,時間為150s,用來帶走反應的副產物和過剩的h2o;步驟(1)~(5)為一個完整沉積過程,循環次數為30次。
實施例4
利用ald設備在高鎳正極材料lini0.5co0.3al0.2o2表面沉積fe2o3薄膜。反應源為h2o和fecl3,沉積溫度為150℃。將高鎳正極材料lini0.5co0.3m0.2o2放入到ald中的反應腔中,沉積過程如下:(1)將反應腔抽真空7s;(2)通入反應鐵源(fecl3)使得腔體壓力達到7mbar;(3)向反應體系中通入n2,時間為140s,用來帶走反應的副產物和過剩的fecl3;(4)繼續向反應腔內通入h2o,當腔體壓力達到7mbar時結束;(5)向反應體系中通入n2,時間為130s,用來帶走反應的副產物和過剩的h2o;步驟(1)~(5)為一個完整沉積過程,循環次數為20次。
實施例5
利用ald設備在高鎳正極材料lini0.6ti0.4o2表面沉積tio2薄膜,反應源為h2o和ticl4,沉積溫度為150℃。將高鎳正極材料lini0.6ti0.4o2放入到ald中的反應腔中,沉積過程如下:(1)將反應腔抽真空5s;(2)通入反應鈦源(ticl4)使得腔體壓力達到5mbar;(3)向反應體系中通入n2,時間為130s,用來帶走反應的副產物和過剩的ticl4;(4)繼續向反應腔內通入h2o,當腔體壓力達到6mbar時結束,(5)向反應體系中通入n2,時間為120s,用來帶走反應的副產物和過剩的h2o;步驟(1)~(5)為一個完整沉積過程,循環次數為25次。
實施例6
利用ald設備在高鎳正極材料lini0.5co0.3mg0.2o2表面沉積tio2薄膜,反應源為h2o和金屬鈦,沉積溫度為150℃。將高鎳正極材料lini0.5co0.3mg0.2o2放入到ald中的反應腔中,沉積過程如下:(1)將反應腔抽真空10s;(2)通入反應鈦源(金屬鈦)使得腔體壓力達到5mbar;(3)向反應體系中通入n2,時間為150s,用來帶走反應的副產物和過剩的金屬鈦;(4)繼續向反應腔內通入h2o,當腔體壓力達到6mbar時結束,(5)向反應體系中通入n2,時間為150s,用來帶走反應的副產物和過剩的h2o;步驟(1)~(5)為一個完整沉積過程,循環次數為30次。
實施例7
將實施例1制備的高鎳正極材料和導電劑炭黑和粘結劑聚四氟乙烯按照一定比例混合,其中正極材料的質量分數比為50%,導電劑的質量分數比為30%,粘結劑的質量分數比為20%,為了調整漿料濃度可加入一定量的nmp(n-甲基吡咯烷酮),混合均勻后涂覆在鋁箔集流體上。
實施例8
將實施例2制備的高鎳正極材料和導電劑天然石墨和粘結劑聚丙烯酸按照一定比例混合,其中正極材料的質量分數比為75%,導電劑的質量分數比為15%,粘結劑的質量分數比為10%,并加入一定量的nmp,混合均勻后涂覆在鋁箔集流體上。
實施例9
將實施例3制備的高鎳正極材料和導電劑石墨烯和粘結劑聚丙烯按照一定比例混合,其中正極材料的質量分數比為99.5%,導電劑的質量分數比為0.1%,粘結劑的質量分數比為0.4%,并加入一定量的nmp,混合均勻后涂覆在鋁箔集流體上。
實施例10
將實施例4制備的高鎳正極材料和導電劑炭黑和粘結劑聚酰胺按照一定比例混合,其中正極材料的質量分數比為50%,導電劑的質量分數比為40%,粘結劑的質量分數比為10%,并加入一定量的nmp,混合均勻后涂覆在鋁箔集流體上。
實施例11
將實施例5制備的高鎳正極材料和導電劑炭黑和粘結劑聚四氟乙烯按照一定比例混合,其中正極材料的質量分數比為40%,導電劑的質量分數比為20%,粘結劑的質量分數比為40%,并加入一定量的nmp,混合均勻后涂覆在鋁箔集流體上。
實施例12
將實施例6制備的高鎳正極材料和導電劑炭黑和粘結劑聚丙烯按照一定比例混合,其中正極材料的質量分數比為99%,導電劑的質量分數比為0.9%,粘結劑的質量分數比為0.1%,并加入一定量的nmp,混合均勻后涂覆在鋁箔集流體上。
實施例13
將實施例7制備的正極極片做成2032型扣式電池,其中,電池殼為不銹鋼材料,負極極片采用鋰金屬,隔膜為celgard2300,電解液為1.0mol/llipf6,溶劑為ec/dmc體系。
實施例14
將實施例8制備的的正極極片做成2032型扣式電池,其中,電池殼為不銹鋼材料,負極極片采用鈉金屬,隔膜為玻璃纖維隔膜,電解液為1.0mol/llipf4,溶劑為emc/dmc體系。
實施例15
將實施例9制備的正極極片做成2032型扣式電池,其中,電池殼為不銹鋼材料,負極極片采用鋰金屬,隔膜為芳綸隔膜,電解液為1.0mol/lliasf6,溶劑為ec/pc體系。
實施例16
將實施例10制備的正極極片做成2032型扣式電池,其中,電池殼為不銹鋼材料,負極極片采用鉀金屬,隔膜為陶瓷涂覆層隔膜,電解液為1.0mol/llicl,溶劑為ec/dmc體系。
實施例17
將實施例11制備的正極極片做成2032型扣式電池,其中,電池殼為不銹鋼材料,負極極片采用鈉金屬,隔膜為無紡布隔膜,電解液為1.0mol/llicl,溶劑為pc/dmc體系。
實施例18
將實施例12制備的正極極片做成2032型扣式電池,其中,電池殼為不銹鋼材料,負極極片采用鉀金屬,隔膜為聚丙烯聚乙烯雙層復合膜,電解液為1.0mol/llibob,溶劑為碳酸乙烯酯。
圖2是未包覆的nca(高鎳正極材料)粉末和本發明實施例1制備的包覆不同厚度薄膜的高鎳正極材料的xrd圖譜(包覆厚度分別為2nm、4nm、6nm),利用xrd可以分析包覆過程中是否引入雜質以及確定包覆物成分。從xrd圖譜中可以看出,本發明利用ald進行表面改性,并沒有引起nca材料的結構變化,所有的明顯的衍射峰對應于α-nafeo2結構,其中分裂明顯的i(006)/i(102)峰和i(018)/i(110)峰說明經過包覆后的材料仍然保持著良好的二維層狀結構。另外,在xrd的圖譜中并沒有出現tio2的衍射峰,主要是因為沉積的厚度在2nm~6nm,tio2的含量較低因此并沒有檢測到tio2的存在。因此,ald并沒有改變nca的晶體結構和成分。
圖3是未包覆的nca和通過包覆進行表面修飾后的nca的sem圖,其中圖3(a)和圖3(b)為包覆前,圖3(c)和圖3(d)為包覆后。從圖中可以看出,包覆前后nca顆粒仍然保持球形形貌,顆粒大小約為5~10μm,整個球形顆粒由初級晶粒構成,初級晶粒為不規則的顆粒,從sem圖中并不能看出包覆層的存在,為了進一步確定包覆層,利用tem進一步分析ald表面包覆層帶來的材料表面結構的變化。
圖4是未包覆的nca和表面包覆如tio2包覆層的nca的tem圖,由于包覆層厚度為4nm,不能夠直接從sem圖中觀察到包覆層的存在,因此利用tem能夠更加清晰的觀察顆粒表面的形貌變化。從圖4(a)中可以看出,未包覆的nca顆粒表面平整且光滑,而利用ald包覆tio2后,顆粒表面粗糙有明顯的包覆層,如圖4(b)所示,包覆層的厚度約為4nm,與ald預設的厚度基本一致,說明利用ald能夠很好控制包覆層的厚度。
為了研究ald沉積包覆層對nca的影響,在1c(180mag-1)的電流密度下對本發明實施例13制備的電池進行恒流充放電測試,電壓范圍為2.8~4.3v,測試溫度為25℃,其測試結果如圖5所示。
圖5(a)是未包覆的nca和ald-2(包覆厚度為2nm)、ald-4(包覆厚度為4nm)、ald-6(包覆厚度為6nm)制備的電池的首圈充放電曲線,其中橫坐標為質量比容量,縱坐標為電壓,從圖中可以看出包覆前后制備的電池的充放電曲線相似,放電電壓平臺均在3.7v左右,且首圈放電容量分別為161.5mahg-1、160.7mahg-1、173.5mahg-1、165.2mahg-1,放電容量并沒有發生較大的變化,首圈效率分別是71.5%、79.3%、78.7%和73.7%。經過包覆后的nca首圈效率均高于未包覆的nca,當包覆層的厚度增加至6nm時首圈效率呈現降低的趨勢。
圖5(b)是未包覆的nca和ald-2、ald-4、ald-6制備的電池的循環性能圖,其中橫坐標為循環圈數,縱坐標為放電比容量。在充放電的循環過程中,在循環的前幾圈中放電容量略微升高,是由材料的活化造成的。從圖中可以看出ald-4的循環性能要優于其他樣品,未包覆的nca在經過100圈充放電循環后,放電容量僅為141.8mahg-1,容量保持率87.8%,ald-2經過100圈充放電循環后,放電容量為155.2mahg-1,容量保持率96.5%,ald-4經過100圈充放電循環后,放電容量為169.0mahg-1,容量保持率97.4%,ald-6經過100圈充放電循環后,放電容量為149.7mahg-1,容量保持率90.6%。從實驗結果可以看出,經過包覆后的nca的循環性能得到了大的提升,特別是當包覆層的厚度為4nm,容量保持率從87.8%提升到97.4%,同時還沒有降低放電容量,但是當包覆層的厚度提高到6nm時,容量保持開始下降,這可能是由于當材料表面的包覆層的厚度太厚會阻礙li+的擴散,因此最佳的包覆層的厚度為4nm。
圖5(c)是未包覆的nca和ald-2制備的電池在不同電流密度下的倍率性能,其中橫坐標為循環圈數,縱坐標為放電比容量。包覆前的nca在0.1c、0.2c、0.5c、1c、2c、5c和10c下的放電容量分別為180.2mahg-1,183mahg-1,174.6mahg-1,166.7mahg-1,157.4mahg-1,136.5mahg-1和103.9mahg-1,可以看出在大倍率的電流密度下,未包覆的nca的電化學性能較差,說明材料的極化較為嚴重,而ald-2在相同的電流密度下放電比容量分別為193.2mahg-1,190.4mahg-1,181.3mahg-1,174.2mahg-1,165.4mahg-1,150.2mahg-1和128.9mahg-1,經過表面包覆后的nca具有良好的倍率性能,這說明包覆層能夠在一定程度上降低材料的極化程度。
圖5(d)是包覆前后的nca在不同倍率下的放電曲線,其中橫坐標為質量比容量,縱坐標為電壓,從圖中也可以看出隨著電流密度的增加,經過包覆后的nca降低了電壓衰減的程度,由此也可以說明nca材料在包覆層作用下,能夠減小材料在充放電過程中的極化問題。
因此,通過高效和良好的循環性能表示經表面包覆能夠提高高鎳正極材料的結構穩定性,從而提高高鎳正極材料的循環性能。
本領域的技術人員容易理解,以上所述僅為本發明的較佳實施例而已,并不用以限制本發明,凡在本發明的精神和原則之內所作的任何修改、等同替換和改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!