一種鋅空氣電池氧化鋅負極材料及其制備方法與流程
本發明涉及一種氧化鋅負極材料及其制備方法,特別涉及一種用于堿性鋅空氣二次電池的氧化鋅負極材料,屬于空氣電池領域。
背景技術:
金屬空氣電池具有容量大、能量密度高的優點。其中,水系鋅空氣電池因具有電池結構簡單、能量密度大、便于存儲、成本低、安全及環保無污染的優點被認為是最有應用潛力的二次金屬空氣電池之一。然而,鋅空氣電池負極由于存在負極產物氧化鋅溶解度大,充電時易產生形變和枝晶生長的問題,致使其放電容量衰減較快,電池循環壽命短,商業化應用受到很大的限制。
為了限制氧化鋅在堿性電解液中的溶解,人們采用支撐電解液或使用堿土金屬氧化物復合氧化鋅阻止氧化鋅溶解,在電解液中加入添加劑來抑制枝晶生長及形變的方法以期望提高鋅負極的循環性能。但是,這些方法的效果均不能令人滿意。表面包覆作為一種常用的材料表面改性手段,在鋰離子電池、鈉離子電池、鋰-硫電池等電極材料的制備方面均有使用,并取得了不錯的效果,這一技術在鋅空氣電池負極材料方面也取得了較好的效果。例如通過在氧化鋅表面包覆sn、bi、ni等金屬化合物可以減少電極添加劑的使用量,改善活性顆粒表面的電流分布,限制放電產物擴散及活性顆粒的電團聚,從而顯著提高鋅負極的循環性能。
雖然表面包覆改善了氧化鋅表面電子的傳輸效果,但一定程度上也阻礙了電解液的輸送,不利于鋅負極的大電流充電;同時,剛性的無機殼結構易受枝晶生長、形變,以及zn/zno晶型轉變產生的體積膨脹作用而破碎崩塌,結構得不到維持,循環壽命較短。
技術實現要素:
針對現有的二次鋅空氣電池負極活性物質表面包覆改性造成的h2o及oh-的擴散受阻,鋅負極大電流充電能力較低,包覆層結構強度不夠,易于破碎崩塌,循環壽命不足的問題,本發明的第一個目的是提供一種在均化活性顆粒表面電流,有效限制放電產物擴散及活性顆粒電團聚的同時,能保證電解液快速穿過表面包覆層到達反應界面,提高表面包覆氧化鋅的大電流充電能力,防止顆粒枝晶生長、形變,以及zn/zno晶型轉變產生的體積膨脹造成的包覆層破碎崩塌,延長鋅空氣電池循環壽命的氧化鋅負極材料。
本發明的第二個目的是提供一種制備如上所述氧化鋅負極材料的方法。
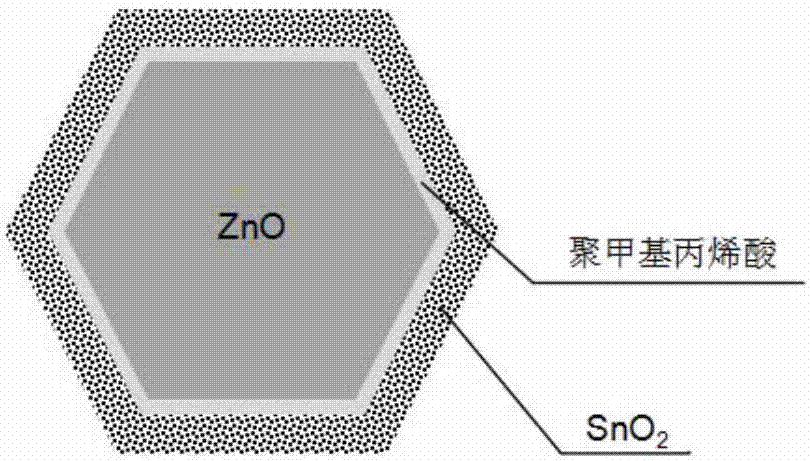
為了實現上述技術目的,本發明提供了一種鋅空氣電池氧化鋅負極材料,所述氧化鋅負極材料具有雙包覆層結構;所述雙包覆層結構為,在氧化鋅與外包覆層之間還均勻分布有內包覆層;所述內包覆層為聚合物包覆層;所述聚合物包覆層中所用聚合物選自耐強堿的聚丙烯酸、聚甲基丙烯酸或丙烯酸和甲基丙烯酸共聚物中的一種;所述外包覆層為氫氧化銦(in(oh)3)和/或二氧化錫(sno2)層。
本發明所述氧化鋅負極材料的雙包覆層總厚度為4~28nm,內包覆層厚度為2~15nm,外包覆層厚度為2~26nm。
本發明還提供了一種制備所述的鋅空氣電池氧化鋅負極材料的制備方法,該方法是:以商業氧化鋅為活性物質,依次在其上沉積耐強堿的親水性聚合物和具有高析氫過電位的金屬的氫氧化物或氧化物。
具體方案包括下述步驟:
步驟一
將氧化鋅粉末超聲分散到去離子水中,加入氨基硅烷偶聯劑,攪拌至少2小時后,固液分離,清洗分離所得固體,干燥,得到表面改性的氧化鋅粉末;
步驟二
將步驟一所得表面改性的氧化鋅粉末加入到聚合物單體溶液中,攪拌均勻后,在60~95℃進行聚合;反應結束后,固液分離,清洗固液分離所得固體,干燥,得到包覆有聚合物層的氧化鋅粉末;所述聚合物單體選自丙烯酸、甲基丙烯酸中的至少一種;
步驟三
將步驟二所得包覆有聚合物層的氧化鋅粉末置于外包覆層反應液中,于40~100℃進行沉積反應;反應結束后,固液分離,清洗固液分離所得固體,干燥,得到鋅空氣電池氧化鋅負極材料;所述外包覆層反應液中含有三價的in和/或六價的sn。
作為優選方案,步驟一中,所述氧化鋅粉末的粒度為0.1-2微米。進一步優選為0.2-0.7微米。
作為優選方案,步驟一中,氨基硅烷偶聯劑的用量為氧化鋅粉末用量的10~100wt%;將氧化鋅粉末超聲分散到去離子水中,加入氨基硅烷偶聯劑后,溶液中氨基類硅烷偶聯劑的濃度為0.0045~0.045mol/l。
作為進一步的優選方案,步驟一中,所述氨基硅烷偶聯劑包括氨丙基三乙氧基硅烷、氨丙基三甲氧基硅烷中的至少一種。
作為優選方案,步驟一中,攪拌2-6小時后,產物經離心分離后,用乙醇和水交替洗去過量偶聯劑,真空干燥,得到表面氨基功能化的氧化鋅粉末。
作為優選方案,步驟二中,聚合物單體的用量為氧化鋅質量的0.43~6%;所述聚合物單體溶液中,聚合物單體的濃度為0.1~1.395mmol/l。
作為優選方案,步驟二中,將步驟一所得表面改性的氧化鋅粉末加入到聚合物單體溶液中,攪拌均勻后,加入引發劑,在60~95℃進行聚合3~8h后;離心分離,用乙醇和水交替洗滌后,真空干燥,得到包覆有聚合物層的氧化鋅粉末;所述聚合物單體選自丙烯酸、甲基丙烯酸中的至少一種;引發劑用量為聚合物單體質量的1.5~4%。所述引發劑優選為過硫酸銨。
作為優選方案,步驟三中,將步驟二所得包覆有聚合物層的氧化鋅粉末置于外包覆層反應液中,于40~100℃進行沉積反應4~24h后;經離心分離、去離子水洗滌、真空干燥,得到鋅空氣電池氧化鋅負極材料;所述外包覆層反應液的溶劑由乙醇和水按體積比1:1-3組成;所述外包覆層反應液中含有in3+和/或sno42-、且外包覆層反應液中in3+和/或sno42-的濃度為0.2~8mmol/l。
作為進一步的優選方案,當in3+由水合硝酸銦(in(no3)3·4.5h2o)提供,sno42-由水合錫酸鈉(na2sno3·4h2o)和/或水合錫酸鉀(k2sno3·3h2o)提供時;其總使用量為所用氧化鋅質量的1.13~61wt%。
作為進一步的優選方案,所述外包覆層反應液中,in3+的濃度為0.9~2.8mmol/l,sno42-的濃度為2~5mmol/l。
作為進一步的優選方案,所述外包覆層反應液中還含有環六亞甲基四胺。作為更進一步的優選方案,所述外包覆層反應液中環六亞甲基四胺的濃度為0.05~0.15mol/l。
與現有技術相比,本發明的技術方案帶來以下有益效果:
(1)本發明的氧化鋅負極材料在金屬無機化合物包覆層內增添了一層適當厚度的親水性聚合物,緩解了單一金屬化合物包覆造成電解液通過包覆層時擴散困難的問題。由于親水性聚合物超強的親水性,電解液中的水及離子自包覆層外通過無機包覆層的阻力得到極大的減小,濃差極化得到降低,因此,鋅負極的大電流充電能力得到提高。
(2)本發明的氧化鋅負極材料采用適當厚度的聚合物-金屬氫氧化物/氧化物雙包覆層復合結構,解決了單一金屬化合物包覆存在的包覆層結構強度低的缺點。由于高分子聚合物的引入,聚合物-金屬氫氧化物/氧化物的復合增強了包覆層的結構強度,提高了其對枝晶生長、形變,以及zn/zno晶型反復轉變產生的體積膨脹的耐受力,從而提高了鋅負極的充放電次數,延長了電池的循環壽命。
(3)本發明氧化鋅負極材料的優選制備方法中,采用硅烷偶聯劑對氧化鋅表面進行改性以及使用環六亞甲基四胺沉積外包覆層,解決了內包覆層包覆不均勻及外包覆層難以包覆的問題。用氨基硅烷偶聯劑對氧化鋅顆粒表面進行氨基功能化,使聚合物均勻地沉積到氧化鋅表面;同時,使用環六亞甲基四胺進行外包覆層的沉積,在減緩無機物的沉積速度,縮小沉積顆粒尺寸的同時,能使外包覆層在聚合物表面順利包覆,從而實現雙包覆氧化鋅活性材料的制備。
總之,本發明采用硅烷偶聯劑和環六亞甲基四胺,成功實現了在氧化鋅顆粒表面進行雙包覆層的均勻沉積。雙包覆層的親水性內包覆層提高了水和離子通過外包覆層的擴散速度,減小了其陰極過程的過電位,同時,聚合物包覆和無機物包覆的復合提高了包覆層的結構強度,防止了包覆層的迅速破壞崩塌,最終提高了氧化鋅負極材料的倍率性能及電池的循環壽命。
附圖說明
圖1本發明柱形氧化鋅負極材料的結構示意圖。
圖2本發明六角片形氧化鋅負極材料的結構示意圖。
具體實施方式
以下具體實施例旨在進一步說明本發明內容,而不是限制本發明權利要求的保護范圍。
實施例1:
氧化鋅負極材料:內包覆層聚丙烯酸層厚度5nm,外包覆層氫氧化銦厚度3nm。
氧化鋅負極材料的制備:將0.5g氧化鋅(粒度為0.2-0.7微米)加入到50ml去離子水中,超聲分散30min,加入0.05g氨丙基三乙氧基硅烷,常溫攪拌5小時。產物經離心分離后,用乙醇和水交替洗滌3次,真空干燥,得到氨基功能化的氧化鋅。然后,將氨基功能化的氧化鋅加入到250ml濃度為0.26mmol/l丙烯酸單體溶液中,超聲分散10min并攪拌2h,加入含量為聚合物單體2wt%的過硫酸銨引發劑于80℃開始聚合反應。反應4h后,將產物離心分離,用乙醇和水交替洗滌3次,得到親水性聚合物包覆的氧化鋅。將聚合物包覆的氧化鋅超聲分散到100ml濃度為0.9mmol/l水合硝酸銦、0.1mol/l環六亞甲基四胺的外包覆層反應液(乙醇/水,v/v=1:3)中,在攪拌的條件下,于80℃加熱進行沉積反應。反應6h后,產物離心分離并用去離子水洗滌3次,真空干燥8h,最終得到聚丙烯酸-氫氧化銦包覆的氧化鋅負極材料。
鋅負極的制備:將氧化鋅負極材料、導電碳、羧甲基纖維素(cmc)、聚四氟乙烯(ptfe)按80:10:3:7的質量配比制成鋅膏漿料,將漿料均勻地涂覆在1cm×1cm的黃銅網上,在通風干燥箱中,80℃干燥5h,并在20mpa下壓片為0.3mm厚。
鋅負極的電化學性能測試:以6mol/lkoh的飽和氧化鋅溶液為電解液,商用氫氧化鎳(ni(oh)2)電極為正極,組裝成模擬電池。將電池以0.2c倍率充電,0.5c倍率放電至1.2v進行活化,5圈后,以1c倍率充電,1c倍率放電至1.2v進行1c/1c倍率充放電循環測試;以5c倍率充電,5c倍率放電至1.2v進行5c/5c倍率充放電循環測試。當放電容量降至初始放電容量60%時中止測試。測試結果見表1。
實施例2:
氧化鋅負極材料:內包覆層聚甲基丙烯酸層厚度5nm,外包覆層二氧化錫厚度5nm。
氧化鋅負極材料的制備:將1g氧化鋅加入到100ml去離子水中,超聲分散30min,加入0.3g氨丙基三乙氧基硅烷,常溫攪拌4小時。產物經離心分離后,用乙醇和水交替洗滌3次,真空干燥,得到氨基功能化的氧化鋅。然后,將0.5g氨基功能化的氧化鋅加入到250ml濃度0.26mmol/l甲基丙烯酸單體溶液中,超聲分散10min并攪拌3h,加入含量為聚合物單體1.5wt%的過硫酸銨引發劑于65。c開始聚合反應。反應7h后,將產物離心分離,用乙醇和水交替洗滌3次,得到親水性聚合物包覆的氧化鋅。將聚合物包覆的氧化鋅超聲分散到100ml濃度為2.1mmol/l水合錫酸鈉、0.1mol/l環六亞甲基四胺的外包覆層反應液(乙醇/水,v/v=1:2)中,在攪拌的條件下,于60℃加熱進行沉積反應。反應9h后,產物離心分離并用去離子水洗滌3次,真空干燥8h,最終得到聚甲基丙烯酸-氧化錫包覆的氧化鋅負極材料。
鋅負極制備及測試按實施例1進行。測試結果見表1。
實施例3:
氧化鋅負極材料:內包覆層丙烯酸甲基丙烯酸共聚物層厚度6nm,外包覆層氫氧化銦厚度10nm。
氧化鋅負極材料的制備:將0.5g氧化鋅加入到50ml去離子水中,超聲分散30min,加入0.25g氨丙基三乙氧基硅烷,常溫攪拌3小時。產物經離心分離后,用乙醇和水交替洗滌3次,真空干燥,得到氨基功能化的氧化鋅。然后,將氨基功能化的氧化鋅加入到250ml濃度0.27mmol/l聚合物單體溶液(丙烯酸單體:甲基丙烯酸單體摩爾比=1:1)中,超聲分散10min并攪拌2h,加入含量為聚合物單體3wt%的過硫酸銨引發劑于85℃開始聚合反應。反應5h后,將產物離心分離,用乙醇和水交替洗滌3次,得到親水性聚合物包覆的氧化鋅。將聚合物包覆的氧化鋅超聲分散到100ml濃度為2mmol/l水合硝酸銦、0.08mol/l環六亞甲基四胺的外包覆層反應液(乙醇/水,v/v=1:2)中,在攪拌的條件下,于90℃加熱進行沉積反應。反應15h后,產物離心分離并用去離子水洗滌3次,真空干燥8h,最終得到聚丙烯酸甲基丙烯酸-氫氧化銦包覆的氧化鋅負極材料。
鋅負極制備及測試按實施例1進行。測試結果見表1。
實施例4:
氧化鋅負極材料:內包覆層聚丙烯酸層厚度5nm,外包覆層二氧化錫厚度23nm。
氧化鋅負極材料的制備:將0.5g氧化鋅加入到50ml去離子水中,超聲分散30min,加入0.5g氨丙基三乙氧基硅烷,常溫攪拌2小時。產物經離心分離后,用乙醇和水交替洗滌3次,真空干燥,得到氨基功能化的氧化鋅。然后,將氨基功能化的氧化鋅加入到250ml濃度0.26mmol/l丙烯酸單體溶液中,超聲分散10min并攪拌3h,加入含量為聚合物單體3.5wt%的過硫酸銨引發劑于80℃開始聚合反應。反應4h后,將產物離心分離,用乙醇和水交替洗滌3次,得到親水性聚合物包覆的氧化鋅。將聚合物包覆的氧化鋅超聲分散到100ml濃度為4.9mmol/l水合錫酸鈉、0.06mol/l環六亞甲基四胺的外包覆層反應液(乙醇/水,v/v=1:2.5)中,在攪拌的條件下,于50℃加熱進行沉積反應。反應23h后,產物離心分離并用去離子水洗滌3次,真空干燥8h,最終得到聚丙烯酸-氧化錫包覆的氧化鋅負極材料。
鋅負極制備及測試按實施例1進行。測試結果見表1。
實施例5
氧化鋅負極材料:內包覆層聚丙烯酸層厚度13nm,外包覆層二氧化錫厚度11nm。
氧化鋅負極材料的制備:將0.5g氧化鋅加入到50ml去離子水中,超聲分散30min,加入0.1g氨丙基三乙氧基硅烷,常溫攪拌3小時。產物經離心分離后,用乙醇和水交替洗滌3次,真空干燥,得到氨基功能化的氧化鋅。然后,將氨基功能化的氧化鋅加入到250ml濃度0.67mmol/l丙烯酸單體溶液中,超聲分散10min并攪拌2h,加入含量為聚合物單體2wt%的過硫酸銨引發劑于75℃開始聚合反應。反應6h后,將產物離心分離,用乙醇和水交替洗滌3次,得到親水性聚合物包覆的氧化鋅。將聚合物包覆的氧化鋅超聲分散到100ml濃度為3.25mmol/l水合錫酸鈉、0.13mol/l環六亞甲基四胺的外包覆層反應液(乙醇/水,v/v=1:2)中,在攪拌的條件下,于70℃加熱進行沉積反應。反應6h后,產物離心分離并用去離子水洗滌3次,真空干燥8h,最終得到聚丙烯酸-氧化錫包覆的氧化鋅負極材料。
鋅負極制備及測試按實施例1進行。測試結果見表1。
對比例1-5:
將0.5g氧化鋅超聲分散到100ml含與實施例相同的金屬鹽的反應液中,在攪拌的條件下,于相應溫度進行沉積反應。一定時間后,產物離心分離并用去離子水洗滌3次,最終得到與實施例相應厚度的金屬氧化物/氫氧化物層包覆的氧化鋅負極材料。
鋅負極制備及測試按實施例1進行。測試結果見表1。
對比例6
其他條件均與實施例1一致,不同之處在于沒有加入環六亞甲基四胺。鋅負極制備及測試按實施例1進行。測試結果見表1。
對比例7
其他條件均與實施例1一致,用甲酰胺替代環六亞甲基四胺。鋅負極制備及測試按實施例1進行。測試結果見表1。
表1各實施例與對比例電化學性能對比表
從實施例1-5和對比例1-5可以看出,利用本發明的雙包覆氧化鋅負極材料制備的鋅負極在容量、1c和5c倍率放電的循環壽命大大優于單一金屬氫氧化物/氧化物包覆的氧化鋅負極材料,效果顯著。從實施例1和對比例6、7可以看出適量的環六亞甲基四胺的加入能顯著提升5c倍率放電的循環壽命以及放電比容量,這大大超出了實驗前的預計。
以上所述僅為本發明的較佳實施例,非用以限定本發明權利要求的范圍,其他運用本發明的精神的等效變化,均應俱屬本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!