一種焦磷酸釩鈉/碳復合正極材料、制備及其應用的制作方法
本發明屬于鈉離子電池領域,具體涉及一種鈉離子電池正極材料焦磷酸釩鈉及其形貌調控與碳包覆的制備方法。
背景技術:
人類社會的生存與發展與能源密切相關,化石能源如煤炭、石油、天然氣等自然資源的普遍應用促進了世界的發展與社會的進步。然而,隨著人類的發展和社會的進步,全世界范圍內能源消耗一直在保持著正的增長。由于地球上的這些礦物能源有限,能源危機便成為了新世紀時代的主題。為了解決該問題,大規模儲能技術成為了一個重要的研究領域。
在眾多儲能技術中,二次電池在能源存儲和轉化方面具有重要意義。鋰離子電池目前占據了便攜式移動電子設備的主要市場,而且正在向混合電動汽車市場方向飛速發展,這將導致鋰的需求量將大大增加,然而地球上鋰的資源非常有限,基于這種現狀和需求,儲量豐富的鈉進入了人們的視野,鈉與鋰位于同一主族,具有相似的化學性質,且鈉元素在地殼中的含量遠遠高于鋰,所以,鈉離子電池是一種非常有前景的二次電池。
目前,基于材料開發成本以及應用前景的考慮,鈉離子電池中研究最多的、最受科研人員關注的當屬鈉離子電池正極材料。目前報道的儲鈉正極材料主要有聚陰離子型化合物、普魯士藍類鈉鹽和過渡金屬氧化物。過渡金屬氧化物有鈷酸鈉、錳酸鈉等,其特點在于容量較高,但是循環性能差,特別是在高電壓下,耐過充性能和熱穩定性能差。普魯士藍類鈉鹽naxmy[fe(cn)6](m=fe,co,ni,cu等)是一類含有變價過渡金屬的配合物,這類化合物具有完整的立方晶型,具有三維的空間結構,存在著大量配位空隙,有利于鈉離子的可逆脫出和嵌入,但這類化合物的容量衰減機制尚不清楚,性能還有待提高。聚陰離子型化合物主要有過渡金屬(焦)磷酸鹽,氟磷酸鹽等;磷酸鹽類化合物常以快離子導體型或橄欖石結構穩定存在,含有較大的離子通道和三維開放性的結構骨架;而且過渡金屬磷酸鹽的框架結構較為穩定,用于正極時充放電過程中,具有較好的循環穩定性和較高的安全性能,但是這類材料的主要缺點是電子電導率低,高倍率放電時容量衰減大。所以如何解決聚陰離子型化合物電子導電率低是研究的重點與熱點,近年來,主要的方法集中在表面改性、粒子摻雜和顆粒納米化。表面改性一般是通過包覆等手段材料表面包覆一層導電率高的材料,比如碳包覆,從而提高材料整體導電性;離子摻雜主要是通過金屬、非金屬離子的摻雜來提高物質的本征導電性;顆粒納米化是通過將材料制備成納米尺寸的微粒,減小電子、離子的遷移來提高導電性。而提高導電性的方法也從單一的包覆、摻雜和納米化發展為多種途徑結合來提高材料的導電性。
技術實現要素:
本發明第一目的在于提供一種焦磷酸釩鈉/碳復合正極材料。
本發明第二目的在于提供一種焦磷酸釩鈉/碳復合正極材料的制備方法,旨在提供一種工藝簡單、重復性好、成本低廉、環境友好的制備方法。
本發明第三目的在于提供所述的正極復合材料的在鈉離子電池領域的應用,旨在通過所述的正極材料,提升鈉離子電池的電化學性能。
一種焦磷酸釩鈉/碳復合正極材料,以多孔焦磷酸釩鈉為核,核的表面包覆有多孔的殼層;所述殼層材料為氮摻雜碳材料。
本發明所述結構的復合正極材料為多孔的納米結構,表面包覆有摻氮碳,很好的解決了焦磷酸釩鈉電子導電性差的缺點;具有優異的電學性能,例如,良好倍率性能和長循環穩定性能。
本發明人還發現,控制所述的復合正極材料的粒徑、殼層厚度、表面孔徑大小等參數,可有助于協同提升材料的電學性能。
作為優選,所述的復合正極材料的粒徑為400-1000nm;進一步優選為400-800nm。
作為優選,比表面積為20-400m2/g;進一步優選為100-300m2/g。
殼層的表面的孔徑小于或等于200nm;進一步優選為40~100nm。
作為優選,殼層厚度為10-100nm;進一步優選為15-65nm。
正極材料的納米孔例如包括小于2nm孔徑的微孔、2nm-50nm孔徑的中孔和50-100nm孔徑的大孔。
本發明還提供了一種所述的焦磷酸釩鈉/碳復合正極材料的制備方法,將包含釩源、氧化劑和含氮碳源單體的溶液進行水熱反應;隨后將水熱產物與鈉源和磷源球磨得到前驅體;將前驅體燒結得所述的復合正極材料。
本發明方法,獨創性地將釩源和含氮碳源單體先進行水熱反應,使得含氮碳源單體聚合、原位生長在釩源微粒表面,得到表面改性的釩源(水熱產物),再將改性后的釩源與鈉源和磷源進行混合,燒結得到所述的復合正極材料。本發明方法工藝簡單、重復性好、成本低廉、環境友好。
本發明制備方法,關鍵在于將釩源和所述的含氮碳源單體優先水熱。此外,配合所述的含氮碳源單體物料的選擇、水熱反應條件、na、v、p元素摩爾比以及燒結過程溫度等參數的控制,可將復合正極材料的顆粒粒徑和碳層厚度、均勻性等控制在更合適的范圍內,進而進一步提升制得的復合正極材料的電學性能。
本發明優選的復合正極材料的制備方法,釩源和含氮碳源單體優先經過水熱反應,使得相應的導電聚合物在釩源微粒上原位生長,再將水熱產物與鈉源和磷源球磨得到前驅體,煅燒、洗滌、干燥后得顆粒均勻的氮摻雜碳包覆的多孔焦磷酸釩鈉微粒。
本發明中,將釩源和含氮碳源單體溶解和/或分散在溶劑中,得所述的的溶液,隨后再進行水熱反應。所述的溶劑可為水、或者和水無限比混溶的溶劑,例如,c1~4的醇或丙酮等。
本發明一種更優選的復合正極材料的制備方法,具體包括以下步驟:
步驟(1):先將一定比例的含氮碳源單體、氧化劑和釩源溶解在乙醇與去離子水的混合介質中,經過水熱反應得到表面原位生長導電聚合物的顆粒均勻的釩源;
步驟(2):將步驟(1)制得的產物與鈉源和磷源球磨、干燥得到前驅體;
步驟(3):前驅體經煅燒、洗滌、干燥后得顆粒均勻的氮摻雜碳包覆的多孔焦磷酸釩鈉微粒。
本發明中,優先進行所述的水熱過程不僅調控了多孔、細小均一的形貌,還使導電聚合物原位生長在釩源上,在后續的燒結過程中抑制了顆粒的生長,有利于實現材料納米化,同時形成均勻的摻氮碳包覆層。摻氮碳包覆層導電性比普通碳層導電性更好,所得產物裝成電池倍率性能也更好。該材料為顆粒均勻的氮摻雜碳包覆的多孔焦磷酸釩鈉微粒,多孔結構和納米級的尺寸大大縮短了電子和粒子的遷移路徑,表面包覆的摻氮碳層又均有良好的電子導電性,很好的解決了焦磷酸鹽倍率性能差的問題。
本發明中,通過水熱法制備顆粒均勻的氮摻雜碳包覆的多孔焦磷酸釩鈉微粒。釩源和含氮碳源單體經過水熱反應,使得相應的導電聚合物在釩源微粒上原位生長,再將水熱產物與鈉源和磷源球磨得到前驅體,煅燒、洗滌、干燥后得顆粒均勻的氮摻雜碳包覆的多孔焦磷酸釩鈉微粒。
本發明中,所述的釩源為釩的氧化物或者其有機、無機鹽;優選為五氧化二釩、偏釩酸銨、乙酰丙酮氧釩、乙酰丙酮釩中的至少一種。
進一步優選,所述的釩源為乙酰丙酮釩、五氧化二釩、偏釩酸銨中的至少一種。
作為優選,所述含氮碳源單體優選為含有氮和碳的水溶性可聚合單體。
進一步優選,所述的含氮碳源單體為苯胺、吡咯、吡啶和多巴胺中的至少一種。
更進一步優選,所述的含氮碳源單體為苯胺、吡咯、吡啶中的至少一種。
作為優選,水熱反應過程中,含氮碳源單體與釩源的摩爾比為0.5-10。該優選的比例范圍內得到的水熱產物顆粒細小均一,且適合導電聚合物的原位生長。
進一步優選,所述的含氮碳源單體與釩源的摩爾比為1-8。
更進一步優選,所述的含氮碳源單體與釩源的摩爾比為1.5-4.5;最優選為1.5~2.3。
作為優選,水熱反應前,溶液中的含氮碳源單體的起始濃度為小于或等于10g/ml。該優選的濃度范圍內制備的水熱產物分散性更好,粒度更均勻。濃度過高、顆粒團聚現象嚴重,濃度過低,則產率過低。
進一步優選,所述的含氮碳源單體的起始濃度為0.01-5g/ml;更進一步優選為0.01-0.5g/ml。
水熱反應過程中,優選添加使含氮碳源單體聚合所需的氧化劑;所述的氧化劑優選為高錳酸鉀、過硫酸鈉、過氧化氫中的至少一種。
氧化劑的投加量可為本領域所熟知的投加量,例如,氧化劑和含氮碳源單體摩爾比小于或等于0.15;優選為0.005-0.15;更進一步優選為0.05-0.1。
作為優選,水熱反應溫度為120-250℃。在該溫度下,含氮碳源單體與釩源更易分散在溶劑中,更利于制得形貌均一性好的改性釩源。
進一步優選,水熱反應溫度為150-200℃。
在所述的優選的水熱溫度下,優選的反應時間為10-30h;進一步優選為10-15h。
本發明人發現,通過所述水熱過程的機制,可使水熱范圍得到的產物的顆粒粒徑、包碳層厚度和均勻性等控制在合適程度,有助于進一步提升最終制得的正極復合材料的電化學性能。
本發明中,將水熱后的前驅體顆粒與鈉源、磷源進行球磨、干燥、煅燒得到顆粒均勻的氮摻雜碳包覆的多孔焦磷酸釩鈉微粒。
所述鈉源優選為可在水溶液中溶解、并可電離釋放出na+的化合物。
作為優選,所述的鈉源為碳酸鈉、醋酸鈉、草酸鈉、焦磷酸鈉和磷酸、磷酸一氫銨、磷酸二氫銨、磷酸一氫鈉、磷酸二氫鈉中的至少一種。
進一步優選,鈉源為碳酸鈉、醋酸鈉、草酸鈉、和磷酸、磷酸一氫鈉、磷酸二氫鈉中的至少一種。
所述磷源優選為可在水溶液中溶解、并可電離釋放出po43-的化合物。
作為優選,所述的磷源為焦磷酸鈉、磷酸、磷酸一氫銨、磷酸二氫銨、磷酸一氫鈉、磷酸二氫鈉中的至少一種。
進一步優選,磷源為磷酸、磷酸一氫鈉、磷酸二氫鈉中的至少一種。
本發明人通過大量研究還發現,將鈉源、釩源和磷源的na、v、p元素摩爾比控制在合適的范圍內,有助于提升制得的復合正極材料的電學性能。
作為優選,鈉源、釩源和磷源按na、v、p元素摩爾比1-8∶1∶0.1-10的比例混合。將鈉源中的na、釩源中的v、磷源中的p的比例在所述的優選范圍內,有助于制得雜質較少,物相較純的焦磷酸釩鈉。各比例未控制在所述優選范圍內,都易產生磷酸釩鈉、焦磷酸鈉、磷酸鈉等雜質。
本發明中,鈉、釩的摩爾比為1-8;釩與磷摩爾比為0.1-10。該范圍內得到的焦磷酸釩鈉雜質較少,物相較純。進一步優選,鈉源、釩源和磷源中,na、v、p元素摩爾比1-5∶1∶0.2-7的比例混合。
更進一步優選,鈉源、釩源和磷源中,na、v、p元素摩爾比1-3∶1∶0.2-3。在該優選比例下,所得焦磷酸釩鈉雜質更少,物相更純,電化學性能更優異。
最優選,na、v、p元素摩爾比2-3∶1∶1.5-2。
本發明方法中,在所述的水熱產物和鈉源、磷源球磨后,經過煅燒得到摻氮碳包覆的多孔焦磷酸釩鈉微粒。
本發明中,作為優選,煅燒(燒結)過程在保護性氣氛下進行。
作為優選,所述的保護性氣氛優選為氮氣、氬氣和氫氬混合氣中至少一種。
本發明人發現,對煅燒過程的溫度進行控制,有助于制得性能優異的正極復合材料。
作為優選,煅燒溫度為500-800℃。在前述的水熱條件和原料選擇、配比的協同下,再配合所述的煅燒溫度,制得的球狀結構的正極材料為多孔的納米級微粒,表面包覆均勻的摻氮碳層,將材料納米化的同時實現了摻氮碳包覆,表現出良好倍率性能。
進一步優選,煅燒溫度為500-700℃。
更進一步優選,所述的煅燒溫度500-650℃;最優選為550-650℃。
煅燒過程的升溫速率為1-10℃/min,降溫速率為1-10℃/min。在該優選的升、降溫速率更有利于保證材料形貌結構的規整性。
進一步優選,煅燒過程的升溫速率為4-8℃/min,降溫速率為2-6℃/min。
在所述的煅燒溫度和升、降溫速率下,優選的保溫時間為10-30h;進一步優選為10-15h。
煅燒后的產物經過洗滌、干燥即制得所述的正極材料。
本發明一種優選的鈉離子電池焦磷酸釩鈉正極材料的制備方法,具體包括以下步驟:
步驟(a):將釩源與含氮碳源單體溶于酒精與去離子水的混合溶劑中,再加入碳源單體聚合所需的氧化劑(高錳酸鉀、過硫酸鈉、過氧化氫中的至少一種),在150-200℃下進行10-15h的水熱反應,得到釩源表面原位生長導電聚合物的前驅體(水熱產物);所述的含氮碳源單體與釩源的摩爾比為1-8;
步驟(b):按na、v、p元素摩爾比1-3∶1∶0.2-3的比例將鈉源、磷源和水熱前驅體(步驟a制得的水熱產物)在150-450rad/min下球磨5-20h;
步驟(c):將步驟(b)制得的產物進行煅燒,煅燒溫度為550-650℃,煅燒時間為10-15h,升溫速率為4-8℃/min;煅燒產物經洗滌、干燥得到摻氮碳包覆的多孔焦磷酸釩鈉微粒。
本發明還包括一種采用所述的制備方法制得的鈉離子電池焦磷酸釩鈉正極材料,所述的材料為氮摻雜碳包覆的焦磷酸釩鈉的多孔微粒。
本發明制得的多孔納米級正極材料在材料納米化的同時實現了摻氮碳包覆,多孔結構提供了大量粒子遷移通道,很好的解決了焦磷酸釩鈉電子導電性差的缺點。將制得的材料作為鈉離子電池正極,可表現出良好倍率性能。
本發明還提供了一種所述的焦磷酸釩鈉/碳復合正極材料的應用,將該正極復合材料用作鈉離子正極材料。
作為優選,所述的應用,將其應用于制備鈉離子電池正極。
本發明的技術方案帶來的有益效果:
1)本發明所述的鈉離子電池正極材料焦磷酸釩鈉電壓平臺可達到4v以上,可以實現鈉離子電池的高能量密度。
2)本發明采用水熱法制備顆粒均勻的多孔焦磷酸釩鈉微粒,水熱過程不僅調控了多孔、細小均一的形貌,還使導電聚合物原位生長在釩源上,在后續的燒結過程中抑制了顆粒的生長,有利于實現材料納米化。
3)水熱過程原位生長的導電聚合物層在后續煅燒過程中形成均勻的摻氮碳包覆層;摻氮碳包覆層導電性比普通碳層導電性更好,所得產物裝成電池倍率性能也更好。
4)該材料為顆粒均勻的氮摻雜碳包覆的多孔焦磷酸釩鈉微粒,多孔結構和納米級的尺寸大大縮短了電子和粒子的遷移路徑,表面包覆的摻氮碳層又均有良好的電子導電性,很好的解決了焦磷酸鹽電子導電率低的問題。
5)本發明的制備方法操作簡單可靠、重復性好、成本低廉、環境友好,而且工藝流程短,易于工業化應用。
附圖說明
【圖1】為實施例1制得的鈉離子電池焦磷酸釩鈉正極材料的掃描電鏡圖;
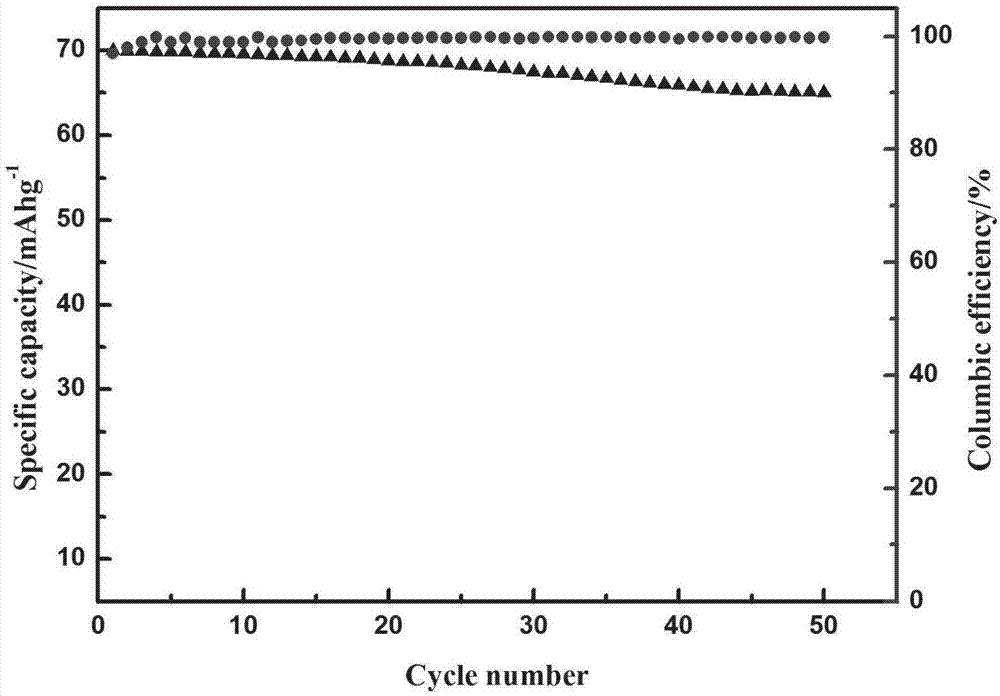
【圖2】為實施例1制得的鈉離子電池焦磷酸釩鈉正極材料的循環圖。
具體實施方式
以下實施例旨在對本發明內容做進一步詳細說明;而本發明權利要求的保護范圍不受實施例限制。
實施例1
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得多孔焦磷酸釩鈉(na7v3(p2o7)4);制得的產品的掃描電鏡圖見圖1;由圖1獲知,通過水熱調控獲得了形貌較好的均一性多孔顆粒。所得復合正極材料的粒徑約為500nm;比表面積約為250m2/g;殼層的表面的孔徑約為40nm;殼層厚度約為15nm。
采用本實施例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能。圖2為本實施例制得的正極材料在電流密度80ma/g的放電比容量與循環效率數據。
由圖2測試結果可知,本例制備的鈉電正極具有良好的電化學性能;在80ma/g(1c)的電流密度下,首圈容量為70mah/g,循環50圈后,仍能保持65mah/g的比容量。
實施例2
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入0.1g過氧化氫(0.003mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為500nm;比表面積約為100m2/g;殼層的表面的孔徑約為40nm;殼層厚度約為40nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為76mah/g,循環50圈后,能保持67mah/g的比容量。
實施例3
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入0.05g過氧化氫(0.0015mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為500nm;比表面積約為100m2/g;殼層的表面的孔徑約為40nm;殼層厚度約為40nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為76mah/g,循環50圈后,能保持67mah/g的比容量。
實施例4
首先稱取3.5g的苯胺(0.045mo1)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,220℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為800nm;比表面積約為300m2/g;殼層的表面的孔徑約為100nm;殼層厚度約為60nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為74mah/g,循環50圈后保持62mah/g的比容量。
實施例5
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應15小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為800nm;比表面積約為300m2/g;殼層的表面的孔徑約為100nm;殼層厚度約為60nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為74mah/g,循環50圈后保持62mah/g的比容量。可見,將水熱過程時間增加至15h,在優選范圍內,材料性能基本不變。
實施例6
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中650℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為500nm;比表面積約為300m2/g;殼層的表面的孔徑約為200nm;殼層厚度約為60nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈容量為76mah/g,循環50圈后,能保持68mah/g的比容量。可見,其他條件不變,煅燒溫度增加至650℃,仍在最優范圍內,材料電化學性能基本不變。
實施例7
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入0.1g過氧化氫(0.003mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結15h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為500nm;比表面積約為100m2/g;殼層的表面的孔徑約為40nm;殼層厚度約為40nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為76mah/g,循環50圈后,能保持67mah/g的比容量。
實施例8
首先稱取3.5g的苯胺(0.045mo1)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入0.1g過氧化氫(0.003mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉4g(0.048mol)、磷酸二氫銨4g(0.0344mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為500nm;比表面積約為100m2/g;殼層的表面的孔徑約為40nm;殼層厚度約為40nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為75mah/g,循環50圈后,能保持67mah/g的比容量。在優選范圍內稍微調整na、v、p的元素摩爾比,產品性能基本不變。
實施例9
首先稱取2.5g(0.037mol)的吡咯、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為550nm;比表面積約為200m2/g;殼層的表面的孔徑約為40nm;殼層厚度約為20nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;測試結果表明,本例制備的鈉電正極倍率性能一般;在80ma/g的電流密度下,首圈容量為67mah/g,循環50圈后,能保持56mah/g的比容量。將苯胺換成吡咯作為碳源,材料結構基本未改變,但采用吡咯制得的材料的電化學性能稍變差。
實施例10
首先稱取2.5g的苯胺(0.034mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為550nm;比表面積約為250m2/g;殼層的表面的孔徑約為100nm;殼層厚度約為100nm。
采用本實施例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為64mah/g,循環50圈后,能保持58mah/g的比容量。結果表明,減小了苯胺與五氧化二釩的摩爾比,倍率性能稍變差。
實施例11
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入0.01g過氧化氫(0.0003mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為500nm;比表面積約為100m2/g;殼層的表面的孔徑約為40nm;殼層厚度約為40nm。
采用本實施例制備的正極材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為76mah/g,循環50圈后,能保持67mah/g的比容量。
實施例12
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于1000ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為550nm;比表面積約為250m2/g;殼層的表面的孔徑約為80nm;殼層厚度約為65nm。
采用本實施例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為68mah/g;循環50圈后,能保持63mah/g的比容量。
實施例13
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,分別在管式爐中500℃和700℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得多孔焦磷酸釩鈉(na7v3(p2o7)4)。500℃下煅燒所得復合正極材料的粒徑約為550nm;比表面積約為200m2/g;殼層的表面的孔徑約為60nm;殼層厚度約為25nm。850℃下煅燒所得復合正極材料的粒徑約為1000nm;比表面積約為150m2/g;殼層的表面的孔徑約為90nm;殼層厚度約為85nm。
采用本實施例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為70mah/g和72mah/g;循環50圈后,分別能保持66mah/g和67mah/g的比容量。
對比例1
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉1.25g(0.015mol)、磷酸二氫銨2.5g(0.0215mol)和干燥后的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得多孔焦磷酸釩鈉(na7v3(p2o7)4)。煅燒所得復合正極材料的粒徑約為650nm;比表面積約為250m2/g;殼層的表面的孔徑約為60nm;殼層厚度約為35nm。
采用本實施例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈容量為57mah/g,循環50圈后,能保持45mah/g的比容量。結果表明,原料鈉、釩和磷元素的比例對產物性能影響較大,超出本發明要求的范圍,制備出的材料電化學性能較差,產物雜相較多。
對比例2
首先稱取0.5g的乙炔(0.019mol)、2g五氧化二釩(0.01mol)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入0.5g過硫酸鈉(0.0021mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,得到最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為650nm;比表面積約為200m2/g;殼層的表面的孔徑約為60nm;殼層厚度約為30nm。
采用本對比例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;測試結果表明,本例制備的鈉電正極電化學性能比實例一的性能差;在80ma/g的電流密度下,首圈比容量為54mah/g,循環50圈后保持45mah/g的比容量。碳層為不含氮的聚乙炔熱解產生的,導電性比聚苯胺熱解得到的摻氮碳更差,電池倍率性能更差。
對比例3
稱取苯胺3.5g(0.045mol)、五氧化二釩2g(0.01mol)、無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)溶于1000ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,180℃下進行水熱反應10小時。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;將干燥產物置于普通瓷舟在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得最終產品焦磷酸釩鈉(na7v3(p2o7)4)。所得復合正極材料的粒徑約為1000nm;比表面積約為250m2/g;殼層的表面的孔徑約為80nm;殼層厚度約為65nm。
采用本對比例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量為61mah/g;循環50圈后,能保持51mah/g的比容量。結果表明,直接將碳源、釩源、鈉源和磷源一步水熱、煅燒,因為沒有進行預包碳,煅燒時材料團聚較嚴重,使得產物粒徑增加,電化學性能變差。
對比例4
首先稱取3.5g的苯胺(0.045mol)、2g五氧化二釩(0.02molv)溶于100ml去離子水和乙醇的混合溶液中,攪拌形成均勻溶液,再加入1g過硫酸鈉(0.0042mol),將所述水溶液倒入聚四氟乙烯反應釜中,然后將所述聚四氟乙烯反應釜放入不銹鋼水熱釜中并密封,最后將所述不銹鋼水熱釜置于均相反應器中,分別在80℃和280℃下進行10小時的水熱反應。將反應后的溶液進行離心處理,然后放置于70℃烘箱里干燥;分別取無水醋酸鈉5g(0.06mol)、磷酸二氫銨5g(0.043mol)和上述干燥的水熱產物球磨10h,轉速300rad/min。球磨后的產物于氬氣氣氛中,在管式爐中600℃高溫燒結10h,產物經去離子水洗滌三次,再用酒精洗滌兩次、干燥,即得多孔焦磷酸釩鈉(na7v3(p2o7)4)。80℃下水熱所得復合正極材料的粒徑約為550nm;比表面積約為200m2/g;殼層的表面的孔徑約為65nm;殼層厚度約為35nm。280℃下水熱所得復合正極材料的粒徑約為600nm;比表面積約為250m2/g;殼層的表面的孔徑約為90nm;殼層厚度約為55nm。
采用本實施例制備的焦磷酸釩鈉材料為工作電極,鈉為對電極,組裝成扣式電池,在80ma/g的電流密度下,測試循環性能;在80ma/g的電流密度下,首圈比容量分別為55mah/g和58mah/g,循環50圈后,分別能保持40mah/g和45mah/g的比容量。80℃和280℃均超出優選溫度范圍120℃-250℃,結果表明,水熱條件特別是水熱溫度對材料性能影響較大,溫度過高或過低都會導致材料形貌不均勻,顆粒粗大,最終導致電池倍率性能變差。
- 還沒有人留言評論。精彩留言會獲得點贊!