一種超薄多孔Co/Fe?N?C納米復合材料及其制備方法和應用與流程
本發明公開了一種超薄多孔co/fe-n-c納米復合材料及其制備方法和應用,屬于電催化劑材料技術領域。
背景技術:
為了解決日益嚴重的能源消耗和環境污染問題,迫切需要探索和開發綠色、高效、可持續的能源或技術來替代傳統基于化石燃料的能源供給和技術體系。金屬-空氣電池,可再生燃料電池,水分解設備由于具有能夠同時存儲和轉換電能而受到廣泛的關注。氧還原反應(orr)是以上設備中一個關鍵的電極反應過程。然而,orr過程受到緩慢動力學的限制。目前,最好的催化orr的催化劑由鉑類金屬組成,這類材料地球儲量少而且昂貴。因此,尋找廉價、高效的電催化劑在這些領域中至關重要。
碳基材料因其分子結構可調,強耐酸性/堿性的獨特優勢,被認為是一種合適的材料來設計催化劑。當納米材料減小尺寸到原子尺度時,由于其原子利用率得到顯著提高,且具有特殊的配位環境,表現出意想不到的性質,所以對于單原子催化材料的制備和應用已經引起了研究者的廣泛關注。
技術實現要素:
發明目的:針對上述技術問題,本發明提供了一種超薄多孔co/fe-n-c納米復合材料及其制備方法和應用。
技術方案:為了達到上述目的,本發明提供了一種超薄多孔co/fe-n-c納米復合材料,所述納米復合材料是以氮摻雜碳為碳基底,結合熔融鹽法,以無機鹽為模板對前驅體進行高溫退火轉化,提高單原子的分散性,從而形成的在碳基底表面和內部均高度分散有單原子co或fe的復合材料。
所述碳基底為超薄多孔的氮摻雜碳,碳源為葉酸或者賴氨酸。
所述復合材料中co顆粒或者fe顆粒為立方相的納米晶。
所述超薄多孔co/fe-n-c納米復合材料的制備方法,包括以下步驟:
將鈷或鐵的無機鹽、葉酸/賴氨酸、甲醇混合,以一定速率升溫反應,調節ph至中性,洗滌,干燥,得到前驅體;然后與無機鹽按照一定摩爾比例充分研磨后,將得到的混合物進行熱處理;最后將產物洗掉無機鹽,超聲分散,刻蝕掉co或fe納米顆粒,洗滌,干燥,即得所述超薄多孔co/fe-n-c納米復合材料。
更具體地,包括以下步驟:
(1)將鈷或鐵的無機鹽、葉酸/賴氨酸、甲醇加入反應器中,以一定速率升溫至50-70℃反應,用naoh溶液調節ph至中性,甲醇洗滌,干燥,得到前驅體;
(2)將步驟(1)所得的前驅體與無機鹽按照一定摩爾比例充分研磨后,將得到的混合物置于管式爐中進行熱處理;
(3)將步驟(2)所得到的產物用去離子水洗掉無機鹽,加入水超聲分散,加入鹽酸刻蝕掉大部分co或fe納米顆粒,洗滌,干燥,得到超薄多孔co-n-c或fe-n-c。
所述步驟(1)中鈷或鐵的無機鹽、葉酸的物質的量比為1:3到3:1,一定速率升溫為:3-20℃/min,反應時間為20min-60min。
所述步驟(2)中無機鹽為nacl、kcl、licl或alcl3中的一種或幾種,其與前驅體的摩爾比為100:1-600:1。
所述步驟(2)中熱處理的溫度為300-900℃,時間為20min-60min。
所述步驟(3)中刻蝕用試劑為鹽酸,其濃度為1mol.l-1-12mol.l-1,刻蝕時間為12-24h。
所述超薄多孔co/fe-n-c納米復合材料可以作為電催化劑應用于電催化氧還原中。
本發明利用葉酸或賴氨酸作為碳源,該兩種碳源具有無毒,且含氮量高等優點,為得到的金屬配合物進一步的碳化提供豐富的碳源和氮源,從而能夠顯著提高催化劑的催化性能。
本發明所述制備方法通過以無機鹽為模板對前驅物高溫退火轉化的方法得到超薄多孔的氮摻雜碳基底上高度分散的單原子co(co-n-c)或fe(fe-n-c)。工藝簡單、可重復性強、適合于批量生產。以氧還原反應(orr)作為探針反應,考察了所得納米材料的電催化性能。該催化劑與和氮摻雜碳(n-c)相比,具有更正的起始電位和更大的極限擴散電流密度,其半波電位可以與商業pt/c相媲美。目前,本發明所述的超薄多孔co/fe-n-c復合物的合成方法以及作為電催化劑應用于電催化orr還未見報道,其在廉價堿性燃料電池領域有著重要的應用前景。
技術效果:相對于現有技術,本發明所述制備方法采用簡單易行的以無機鹽為模板對前驅物高溫退火轉化的方法,合成超薄多孔的氮摻雜碳基底上高度分散的單原子co或fe的復合物,所述方法工藝簡單、可重復性強,通過調節配合物和無機鹽的量來調節產物量,適合批量生產。
附圖說明
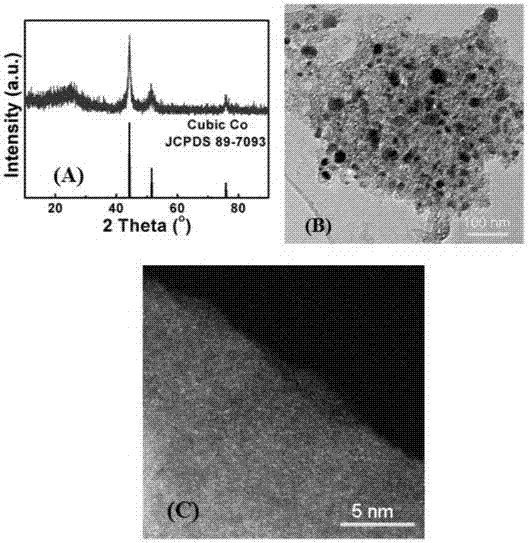
圖1:本發明所述超薄多孔co-n-c復合物的xrd圖譜;
圖2:為本發明所述超薄多孔co-n-c復合物的xps總譜;
圖3:(a)為本發明所述超薄多孔co-n-c復合物的tem圖,標尺為50nm;(b)為本發明所述超薄多孔co-n-c復合物的hrtem圖,標尺為10nm;(c)為本發明所述超薄多孔co-n-c復合物的haadf-stem圖;
圖4:本發明所述超薄多孔co-n-c復合物的orr性能測試圖;
圖5:本發明所述超薄多孔fe-n-c復合物的tem圖。
圖6:(a)為對比例1所述無nacl為模板的co/co-n-c復合物的xrd圖;(b)為對比例1所述無nacl為模板的co/co-n-c復合物的tem圖;(c)為對比例1所述無nacl為模板的co/co-n-c復合物的haadf-stem圖。
具體實施方式
下面通過具體實施例對本發明所述的技術方案給予進一步詳細的說明,但有必要指出,以下實施例只用于對發明內容的描述,并不構成對本發明保護范圍的限制。#
實施例1超薄多孔co-n-c復合物的制備
室溫下,在潔凈,干燥的250ml三頸燒瓶中,加入1.0mmol葉酸(fa)和1.0mmolcocl2·6h2o加入到甲醇中,以3℃/min速率升溫至60℃保溫30min調節ph至中性,得到穩定沉淀,用甲醇洗滌,冷凍干燥。取一定量上述樣品與nacl充分研磨(無機物與前驅體的摩爾比為300:1),在ar或n2等惰性氣氛下,900℃保溫20min后自然冷卻至室溫。用超純水洗掉nacl,將得到的黑色沉淀轉移到三頸燒瓶中,加入15ml濃度為1moll-1的稀鹽酸處理24h后,用超純水洗至中性,冷凍干燥,收集樣品。
采用xrd和xps測試對制得的超薄多孔co-n-c復合物組份進行分析(如圖1、2)。圖1中的衍射峰與標準卡片相對應,證明沒有立方相的co的存在。圖2的xps總譜中有c,n,o的存在,少量的co被檢測到,o可能來自空氣或者表面吸附氧。
采用tem、hrtem和haadf-stem圖(圖3(a)、3(b)、3(c))分別對得到的超薄多孔co-n-c復合物形貌進行分析,從圖3(a)圖中可以看到本發明的超薄多孔co-n-c復合物主要以三維多孔的超薄c為主體,局部有co納米顆粒的存在。從圖3(b)的htem圖中可以看到c上沒有co顆粒的存在,但從3(c)的haadf-stem中可以看到高度分散的單原子co。
實施例2co-n-c的制備
室溫下,在潔凈,干燥的250ml三頸燒瓶中,加入1.5mmol葉酸(fa)和0.5mmolcocl2·6h2o加入到甲醇中,以20℃/min速率升溫至50℃保溫60min,調節ph至中性,得到穩定沉淀,用甲醇洗滌,冷凍干燥。取一定量上述樣品與kcl+licl充分研磨(無機物與前驅體的摩爾比為400:1),在ar或n2等惰性氣氛下,300℃保溫60min后自然冷卻至室溫。用超純水洗掉kcl+licl,將得到的黑色沉淀轉移到三頸燒瓶中,加入5ml濃度為12moll-1的稀鹽酸處理24h后,用超純水洗至中性,冷凍干燥,收集樣品。
采用xrd,tem等對得到的產物進行表征,得到的仍是超薄多孔co-n-c復合物。
實施例3fe-n-c的制備
室溫下,在潔凈,干燥的250ml三頸燒瓶中,加入1mmol葉酸(fa)和1mmolfecl3·6h2o加入到甲醇中,以12℃/min速率升溫至70℃保溫20min調節ph至中性,得到穩定沉淀,用甲醇洗滌,冷凍干燥。取一定量上述樣品與nacl充分研磨(無機物與前驅體的摩爾比為100:1),在ar或n2等惰性氣氛下,600℃保溫40min后自然冷卻至室溫。用超純水洗掉nacl,將得到的黑色沉淀轉移到三頸燒瓶中,加入10ml濃度為6moll-1的稀鹽酸處理12h后,用超純水洗至中性,冷凍干燥,收集樣品。
采用xrd,tem等對得到的產物進行表征,結果如圖5所示,得到的仍是超薄多孔fe-n-c復合物。
實施例4fe-n-c的制備
室溫下,在潔凈,干燥的250ml三頸燒瓶中,加入1mmol賴氨酸和1mmolfecl3·6h2o加入到甲醇中,以15℃/min速率升溫至70℃保溫20min調節ph至中性,得到穩定沉淀,用甲醇洗滌,冷凍干燥。取一定量上述樣品與nacl充分研磨(無機物與前驅體的摩爾比為600:1),在ar或n2等惰性氣氛下,600℃保溫40min后自然冷卻至室溫。用超純水洗掉nacl,將得到的黑色沉淀轉移到三頸燒瓶中,加入10ml濃度為6moll-1的稀鹽酸處理12h后,用超純水洗至中性,冷凍干燥,收集樣品。
采用xrd,tem等對得到的產物進行表征,結果可得,得到的仍是超薄多孔fe-n-c復合物。
實施例5超薄多孔co-n-c復合物作為電催化劑在orr中的應用
超薄多孔co-n-c復合物作為電催化劑催化orr的測試方法如下:稱取2.5mg的超薄多孔co-n-c復合物,溶于0.5ml水、0.5ml乙醇和20μl萘酚的混合溶液中,溶液的濃度為2.5mgml-1,超聲分散均勻后,取8μl上述溶液,將其滴加在干凈的旋轉環盤玻碳電極上,干燥后,重復上述操作一次,干燥后即可用于電化學測試。
對于orr反應,首先在n2飽和的0.1mkoh的溶液中進行循環伏安測定。待其穩定后,將氣體換成o2,通入該電解液中,同樣進行循環伏安測試,待其穩定后進行不同轉速下的極化曲線的測定。
對于orr反應,在o2飽和的0.1mkoh的溶液中進行循環伏安測定,待其穩定后,進行極化曲線測定本發明所述的超薄多孔co-n-c復合物電催化orr性能測試,結果如圖4所示,其起始電位和半波電位分別為0.90v和0.84v(vs.rhe)催化性能優于對應的氮摻雜碳和co/c。按照實施例1方法,將葉酸替換為苯胺,所得復合物進行上述性能測試,結果可得,其起始電位和半波電位分別為0.85v和0.76v(vs.rhe)催化性能均顯著差于實施例1所得復合物的催化性能。
綜上所述,超薄多孔co-n-c復合物表現出優異的電催化orr性能,有望作為一種廉價,高效的催化劑應用于堿性燃料電池領域。
對比例1co-n-c的制備
室溫下,在潔凈,干燥的250ml三頸燒瓶中,加入1mmol葉酸(fa)和1mmolcocl2·6h2o加入到甲醇中,60℃保溫30min調節ph至中性,得到穩定沉淀,用甲醇洗滌,冷凍干燥。取一定量上述樣品在ar或n2等惰性氣氛下,800℃保溫30min后自然冷卻至室溫,收集樣品。
采用xrd(圖6a),tem圖(圖6b)和haadf-stem圖(圖6c)等對得到的產物進行表征,得到的仍為常規的co-n-c,但顆粒聚集嚴重,碳的堆疊程度嚴重。表明nacl在其中起到“溶劑”的作用,使顆粒和碳層分散均勻。
對比例2co-n-c的制備
室溫下,在潔凈,干燥的250ml三頸燒瓶中,加入0.5mmoll-賴氨酸和1.5mmolcocl2·6h2o加入到甲醇中,60℃保溫30min調節ph至中性,得到穩定沉淀,用甲醇洗滌,冷凍干燥。取一定量上述樣品在ar或n2等惰性氣氛下,800℃保溫30min后自然冷卻至室溫,收集樣品。將得到的黑色沉淀轉移到三頸燒瓶中,加入5-15ml適當濃度的稀鹽酸處理24h后,用超純水洗至中性,冷凍干燥,收集樣品。
采用xrd,tem圖和haadf-stem圖等對得到的產物進行表征,得到的仍為常規的co-n-c。
- 還沒有人留言評論。精彩留言會獲得點贊!