鋰空氣電池陰極用鈣鈦礦結構硫化物催化劑材料及其制備方法與流程
本發明屬于鋰空氣電池領域,具體涉及一種陰極用鈣鈦礦結構硫化物催化劑材料及其制備方法。
背景技術:
鋰空氣電池是一種新型的儲能體系,具有成本低、清潔環保、可循環使用等優點,同時還擁有可與汽油相媲美的高理論能量密度(11400wh·kg-1),在未來極有可能取代鋰離子電池而成為更高效的能量存儲與轉化系統,因而近年來受到了廣泛的關注。鋰空氣電池的負極活性物質為自然界最輕的金屬元素鋰、正極活性物質為空氣中的氧氣。在電池的放電(氧還原反應)過程中,通過金屬鋰與氧氣反應生成過氧化鋰附著于多孔陰極表面完成由化學能到電能的轉換;在充電(析氧反應)過程中,通過過氧化鋰分解為金屬鋰和氧氣實現由電能到化學能的轉換。
經過近十年來的發展,鋰空氣電池的性能雖已取得了長足的進步,但目前仍面臨著能量轉換效率低、倍率性能差、循環壽命短等問題。在諸多影響鋰空氣電池性能的因素中,陰極緩慢的氧還原反應(orr)和析氧反應(oer)動力學起著關鍵的作用。因此,需要在陰極使用具有雙功能催化作用的催化劑來加快電極反應、改善電池性能。目前所使用的正極催化劑材料主要包括碳材料、貴重金屬、過渡金屬及其氧化物等。其中,碳材料易腐蝕失活并生成副產物;貴金屬資源有限、價格昂貴;過渡金屬及其氧化物的催化性能難以滿足要求。所有這些都大大地限制了鋰空氣電池的性能、發展和規模應用。因此,廉價、高效的非貴金屬催化劑的成功開發對鋰空氣電池已成為亟待解決的迫切任務。
近年來,硫化物在儲能領域的應用逐漸受到關注。鈣鈦礦結構硫化物作為一類具有特殊結構的硫化物,具有原料儲量豐富、價格低廉、結構穩定、離子導電率高等優點。雖然部分鈣鈦礦結構硫化物,如bazrs3和srzrs3等,已被證明可作為高效的光伏材料應用于太陽能儲能系統(advancedmaterials,29(2017)1604733),但將鈣鈦礦結構硫化物材料用作鋰空氣電池的陰極催化劑至今尚未見報道。鈣鈦礦結構硫化物的分子式通常表示為abs3,a位和b位分別是一種或多種結構的陽離子,a位和b位陽離子的總的化學價等于+6價。通常,a位陽離子比b位陽離子的離子半徑大,a位典型代表包括ca、sr、ba、pb、sn等金屬元素的陽離子,b位典型代表包括ti、zr、hf等金屬元素的陽離子,此外還可能包含非金屬陽離子,例如ch3nh3+、(nh3nh3)2+等。b位離子與s2-形成正八面體結構的bs6,b處于八面體的中心,s占據八面體的各個頂點;a位離子占據由八個bs6形成的立方八面體的空穴位。此外,雖然鈣鈦礦結構硫化物的分子通式寫為abs3,但a位和b位的陽離子可以被其他離子部分取代形成更為復雜的鈣鈦礦結構,進一步擴充鈣鈦礦結構硫化物材料的種類,相應的材料性能也能滿足更多的要求。
技術實現要素:
本發明的目的就是為了克服上述現有技術存在的缺陷而提供一種方法簡單、重復性好的鋰空氣電池陰極用鈣鈦礦結構硫化物催化劑材料及其制備方法。
本發明的目的可以通過以下技術方案來實現:一種鋰空氣電池陰極用鈣鈦礦結構硫化物催化劑材料,其特征在于,該材料的分子通式為abs3,其中,a為金屬或非金屬陽離子,b為金屬或非金屬陽離子,s為硫離子,其中,a和b的總的化學價等于+6價,所述材料為鈣鈦礦結構硫化物。
所述的金屬包括ca、sr、ba、pb、sn、ti、zr或hf,所述的非金屬陽離子包括ch3nh3+或(nh3nh3)2+。
所述的a為同一種陽離子,或者該陽離子被其他一種或多種陽離子部分取代。
所述的b為同一種陽離子,或者該陽離子被其他一種或多種陽離子部分取代。
一種鋰空氣電池陰極用鈣鈦礦結構硫化物催化劑材料的制備方法,其特征在于,制備方法包括溶劑熱法、abx3硫化法,其中x為非硫元素,或高溫固相合成法。
所述的溶劑熱法包括以下步驟:按照化學計量比稱取a源、b源和硫源并均勻分散于溶劑中,然后轉移至高壓反應釜中加熱到100-300℃,保溫5-50h,之后自然冷卻到室溫,經反復洗滌并干燥后得到催化劑;
其中,a源、b源包括a和b的金屬單質、a和b的氯化物、碳酸鹽、硝酸鹽、醋酸鹽或金屬有機化合物;硫源包括硫單質、硫脲及硫代乙酰胺;溶劑包括去離子水或有機溶劑。
所述的abx3硫化法由相應的abx3在高溫下與硫源反應制得,主要包括以下步驟:將相應的abx3粉末置于高溫反應容器中,引入硫源,升溫至300-1600℃,保溫2-300h,隨后自然冷卻得到催化劑;
其中,x包括s的同族元素或鹵素;硫源包括硫單質、h2s或cs2。
所述的高溫固相合成法包括以下步驟:將a源、b源和硫源充分研磨后冷壓,之后裝入高溫反應器中,加熱至300-1500℃并保溫2-84h,反應結束后冷卻至室溫。將所得產物研磨后在惰性氣氛下于400-1500℃下進行1-40h的熱處理,得到催化劑;
其中,a源、b源包括但a和b的金屬單質、氧化物、硫化物、鹽或金屬有機化合物;硫源包括硫單質、a和b硫化物、或硫脲。
與現有技術相比,本發明公開的鋰空氣電池陰極用鈣鈦礦結構硫化物催化劑材料,相較于已經采用的鈣鈦礦結構氧化物,因為其中硫原子的電負性比氧小,相應的鈣鈦礦材料中的八面體結構bs6比bo6的畸變程度小,有利于保持理想的立方鈣鈦礦結構;硫的原子半徑比氧大,使得其相應鈣鈦礦結構中過渡金屬原子d軌道分裂能減小,晶胞體積增大,晶胞中的原子密度減小;o2p價電子軌道和過渡金屬d導帶軌道的能隙高于s3p和過渡金屬d導帶軌道的能隙。這些特性使得鈣鈦礦結構硫化物比氧化物具有更高的導電性、更好的鋰離子和氧離子傳遞性,同時也使得其催化性能更容易發揮,從而作為鋰空氣電池陰極催化劑具有更優異的性能。
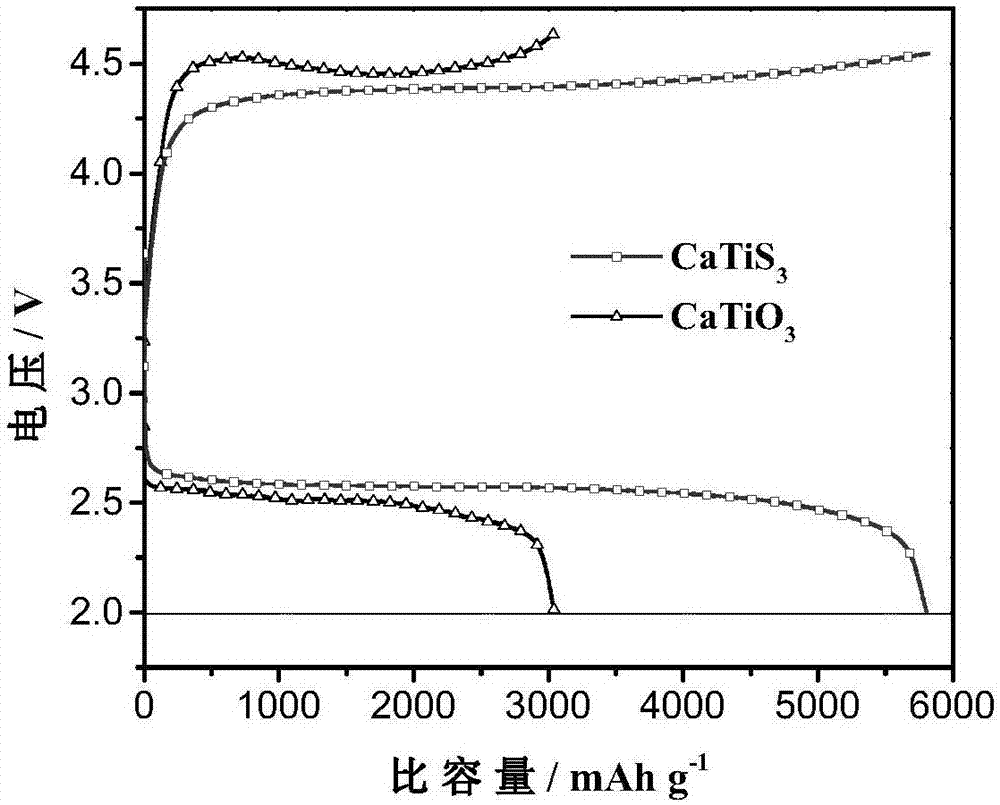
以實施例1制得的鈣鈦礦結構硫化物催化劑材料catis3為例,基于其作為陰極催化劑的鋰空氣電池的放電比容量達到了5805mah·g-1,明顯高于以比較例1制得的鈣鈦礦氧化物材料catio3為催化劑的鋰空氣電池;放電電壓平臺約為2.65v,充電電壓平臺約4.30v,相較于以比較例1制得的鈣鈦礦氧化物材料catio3為催化劑的鋰空氣電池,放電電壓提高了150mv,充電電壓降低了200mv,有效地緩減了電池充放電過程中的極化現象,從而提高了鋰空氣電池的能量轉換效率。此外,基于實施例1制得的catis3的鋰空氣電池的循環性能也較基于比較例1制得的catio3的電池得到了明顯的改善。
附圖說明
圖1是分別基于實施例1和比較例1所得鈣鈦礦結構材料的鋰空氣電池的充放電曲線;
圖2是分別基于實施例1和比較例1所得鈣鈦礦結構材料的鋰空氣電池的循環性能曲線。
具體實施方式
下面結合附圖和具體實施例對本發明進行詳細說明。
鋰空氣電池的電極制備、電池裝配和電化學性能測試方法如下:
電極制備:
利用實施例或比較例所得的材料作為鋰空氣電池陰極催化劑。首先將質量比為6:3:1的superp、催化劑和聚四氟乙烯加入到30ml無水乙醇中,磁力攪拌4h。之后將圓片狀泡沫鎳作為集流體,采用噴涂法將混合均勻的懸濁液負載于集流體上制成電極,材料負載量約為0.5mg·cm-2。
電池裝配:
鋰空氣電池的組裝采用自制的改進型swagelock模具,具體步驟為:以金屬鋰片為負極、玻璃纖維為隔膜、雙三氟甲烷磺酰亞胺鋰的四乙二醇二甲醚溶液(1mlitfsi/tedme)為電解液。首先取金屬鋰片置于模具負極正中央,然后將電解液浸泡過的玻璃纖維隔膜放置于鋰片上,之后將正極片放置于隔膜上,負載有正極材料的一側緊貼隔膜,隨后旋入聚四氟乙烯隔套,加入泡沫鎳并旋入正極殼體擰緊。上述鋰空氣電池的組裝操作在水和氧的含量均低于0.1ppm的手套箱中完成。
電化學性能測試:
室溫下,將裝配好的鋰空氣電池密封于一個大氣壓下的高純氧氣氛中,測試前將電池在氧氣氣氛中靜置4h。電池放電截止電壓設置為2.0v,之后進行等容量的充電過程。電池的充放電電流密度為200ma·g-1。比容量和電流密度基于陰極所用碳材料superp的質量進行計算。充放電測試系統為武漢藍電電子有限公司生產的電池充放電設備(landct2001a)。
實施例1:catis3
稱取0.01molca(no3)2、0.01molti(no3)4、0.05mol硫脲及5mg碘單質(i2)并充分溶解于40ml去離子水中,超聲15min,將混合均勻的溶液放入50ml的高壓反應釜中,加熱至300℃保溫10h,然后自然冷卻至室溫,最后將所得產物用去離子水反復洗滌并于100℃真空干燥12h得到催化劑catis3。
以實施例1所得材料為陰極催化劑的鋰空氣電池表現出良好的性能(附圖1、2)。從圖1可以看出,基于其作為陰極催化劑的鋰空氣電池的放電比容量達到了5805mah·g-1,明顯高于以比較例1制得的鈣鈦礦氧化物材料catio3為催化劑的鋰空氣電池;放電電壓平臺約為2.65v,充電電壓平臺約4.30v,相較于以比較例1制得的鈣鈦礦氧化物材料catio3為催化劑的鋰空氣電池,放電電壓提高了150mv,充電電壓降低了200mv,有效地緩減了電池充放電過程中的極化現象,從而提高了鋰空氣電池的能量轉換效率。此外,從圖2可以看出,基于實施例1制得的catis3的鋰空氣電池的循環性能也較基于比較例1制得的catio3的電池得到了明顯的改善。
實施例2:casns3
稱取1.110gcacl2、3.506gsncl4·5h2o、2.2836g硫脲及10mg碘單質(i2)并溶解于80ml去離子水中,超聲30min,將混合均勻的溶液放入100ml的高壓反應釜中,加熱至100℃保溫35h,然后自然冷卻至室溫,最后將所得產物用去離子水反復洗滌并于80℃真空干燥24h得到催化劑casns3。
實施例3:bazrs3
稱取1.3067gba(no3)2、1.9661gzr(no3)4·3h2o、1.1270g硫代乙酰胺充分溶解于40ml去離子水與乙醇混合溶液中(體積比3:1),超聲15min,將混合均勻的溶液放入50ml的高壓反應釜中,加熱至150℃保溫50h,然后自然冷卻至室溫,最后將所得產物用去離子水反復洗滌并于120℃真空干燥8h得到催化劑bazrs3。
實施例4:pbsns3
稱取0.01molpbcl2、0.01molsncl4·5h2o,0.03mol硫代乙酰胺及0.01mol鹽酸并溶解于150ml去離子水與乙二醇混合溶液(體積比4:1)中,超聲30min,將混合均勻的溶液放入180ml的高壓反應釜中,加熱至200℃保溫10h,然后自然冷卻至室溫,最后將所得產物用去離子水反復洗滌并于100℃真空干燥12h得到催化劑pbsns3。
實施例5:cazrs3
按照化學計量比1:1:3稱取0.05molcacl2、0.05molzr(no3)4、0.15mol硫粉及0.01mol硝酸并溶于80ml去離子水中,超聲30min,將混合均勻的溶液放入100ml的高壓反應釜中,加熱至180℃保溫5h,然后自然冷卻至室溫,最后將所得產物用去離子水反復洗滌并于100℃真空干燥12h得到催化劑cazrs3。
實施例6:cazrs3
稱量50mmolcazro3放于瓷舟中,置于管式爐中,在氬氣氣氛下以15℃min-1的速度升溫,溫度升至100℃時開始用鼓泡器引入h2s氣體置換氬氣,溫度升至300℃時保溫300h。之后以15℃min-1的速度降溫至100℃,停止通h2s,重新通入氬氣并保溫2h,之后冷卻至室溫得到催化劑cazrs3。
實施例7:catis3
稱量30mmolcatio3放于瓷舟中,置于管式爐中,在氮氣氣氛下以5℃min-1的速度升溫,溫度升至800℃時開始用鼓泡器引入h2s氣體置換氮氣,溫度升至1600℃時保溫2h。之后以5℃min-1的速度降溫至800℃,停止通h2s,重新通入氮氣并保溫2h,之后冷卻至室溫得到催化劑catis3。
實施例8:srzrs3
稱量0.1molsrzrse3放于瓷舟中,置于管式爐中,在氬氣氣氛下以10℃min-1的速度升溫,溫度升至400℃時開始用鼓泡器引入cs2氣體置換氬氣,溫度升至900℃時保溫50h。之后以10℃min-1的速度降溫至400℃,停止通cs2,重新通入氬氣并保溫2h,之后冷卻至室溫得到催化劑srzrs3。
實施例9:bazrs3
稱量10gbazrse3放于瓷舟中,置于管式爐中,在氮氣氣氛下以15℃min-1的速度升溫,溫度升至400℃時開始用鼓泡器引入cs2氣體置換氮氣,溫度升至700℃時保溫100h。之后以15℃min-1的速度降溫至800℃,停止通cs2,重新通入氮氣并保溫2h,之后冷卻至室溫得到催化劑bazrs3。
實施例10:bazrs3
分別稱取8.470gbas和12.306gzrs2,將其混合均勻后球磨并冷壓,然后置于石英管中,抽真空至壓強小于7×10-7torr并火焰密封,再以5℃min-1的速率加熱至800℃并保溫30h,之后冷卻至室溫。將所得產物研磨后在氮氣氛下于400℃熱處理40h,得到催化劑bazrs3。
實施例11:cazrs3
稱量7.214gcas和24.613gzrs2,充分研磨后冷壓,之后裝入石英管中,抽真空并密封。將體系加熱至300℃并保溫84h,反應結束后冷卻至室溫。將所得產物研磨后在氬氣氛下于1500℃熱處理1h,得到催化劑cazrs3。
實施例12:cahfs3
稱量cas和hfs2各0.1mol,充分研磨后冷壓,之后裝入石英管中,抽真空并密封。將體系加熱至1500℃并保溫2h,反應結束后冷卻至室溫。將所得產物研磨后在氬氣氛下于1000℃熱處理20h,得到催化劑cahfs3。
實施例13:latis3
稱取18.7003gla2s3、4.8097gs粉和4.7877g金屬ti粉,混合均勻后置于石英管中密封,升溫至900℃,加壓至3.5gpa,然后恒溫恒壓100h。將產物收集起來并研磨后于同樣的溫度、壓力下處理得到催化劑latis3。
實施例14:lacrs3
稱取0.05molla2s3、0.15mols粉和0.1mol金屬cr粉,混合均勻后置于石英管中密封,升溫至1100℃,加壓至2.5gpa,然后恒溫恒壓50h。將產物收集起來并研磨后于同樣的溫度、壓力下處理得到催化劑lacrs3。
比較例1:catio3
稱取0.01molca(no3)2和0.01molti(no3)4,充分溶解于50ml5%的稀硝酸溶液中,以koh作為沉淀劑加入溶液得到ca與ti的共沉淀物,將沉淀過濾并洗滌后與40ml10%的koh溶液同時放入50ml的高壓反應釜中,加熱至200℃保溫10h,然后自然冷卻至室溫,將所得產物用去離子水反復洗滌并于100℃真空干燥12h得到催化劑catio3。
需要說明的是,本發明不限于上述實施例,任何熟悉本技術領域的技術人員在本發明揭露的技術范圍內,可輕易想到的變化或替換,都應涵蓋在本發明的保護范圍之內,因此,本發明的保護范圍應該以權利要求的保護范圍為準。
- 還沒有人留言評論。精彩留言會獲得點贊!