硅碳復合材料及其制備方法和應用與流程
本發明涉及電池技術領域,尤其涉及一種硅碳復合材料及其制備方法和應用。
背景技術:
鋰離子電池作為一種環境友好的化學能源,具有體積小、能量密度高、循環壽命長等優點,被認為是一項非常有前景的儲能方向。目前鋰離子電池已經被普遍使用在3c領域,在hev和ev領域也得到越來越大規模的應用。同時,隨著通信技術的快速發展和各國政府對ev的大力推進,對鋰離子電池能量密度、功率密度和循環壽命的要求也越來越高。相對于正極材料較為成熟的研究,傳統的碳負極材料由于其理論容量較低而成為限制電池容量的制約因素。理想的負極材料應具有成本低、安全性高、能量密度高、循環壽命長的特性。硅基材料是一種很有潛力的負極材料。硅儲量豐富,安全性高,且具有比傳統的碳負極(372mahg-1)高10倍、比鈦酸鋰負極(175mahg-1)高20倍的理論比容量(4200mahg-1)。此外,硅基負極對鋰的電位適中,其起始電位為0.3~0.4vvs.li/li+,這種適中的電位能避免像石墨,電極(0.05vvs.li/li+)一樣發生鋰沉積,影響安全性能;同時也避免了發生如鈦酸鋰電池(1.5vvs.li/li+)的能量損失。盡管有以上優點,硅與鋰合金化/去合金化時巨大的體積變化(高達300%)會引起硅的破裂和粉碎,導致電接觸的損失和最終的容量衰減。通常能通過降低硅材料尺寸和將硅與其他材料復合來達到改進硅基材料循環性能的目的。將硅的尺寸降至納米級后與碳質材料進行復合化,一方面能利用碳材料的高導電性提高復合材料的電導率,另一方面也能作為基質緩沖硅基材料的體積變化。但是納米硅的表面活性較高,易發生團聚,導致硅材料不能很好地分散在碳材料中。現有的方法很難使硅分布于碳之中或均勻包覆在載體上,仍然不能完全避免硅在可逆充放電過程中結構的緩慢破壞。
針對相關技術中的問題,目前尚未提出有效的解決方案。
技術實現要素:
針對相關技術中的上述技術問題,本發明提出一種具有較高的導電性能兼具較為優異的循環穩定性的硅碳復合材料。還提供了該硅碳復合材料的制備方法,以聚合物熱解后形成有機碳包覆在石墨、碳酸鋰、納米硅的表面,起到粘結納米硅與石墨的作用,同時碳包覆減少了球磨后材料的表面積,降低了副反應的發生,此外碳材料也提高了納米硅的導電率。因此,本發明的硅碳復合材料可應用于制備鋰離子電池。
為實現上述技術目的,本發明的技術方案是這樣實現的:
一種硅碳復合材料,包括納米硅、石墨和碳酸鋰,所述納米硅、石墨和碳酸鋰通過聚合物包覆后,經碳化得到硅碳復合材料。
做為一個總的技術構思,本發明還提供了一種鋰離子電池負極材料的制備方法,其特征在于,包括以下步驟:
s1、將石墨、碳酸鋰、納米硅混合得到碳酸鋰/硅/石墨混合物;
s2、將所述碳酸鋰/硅/石墨混合物分散到聚合物溶液中,經干燥得到前驅體材料;
s3、將所述前驅體材料進行碳化得到碳硅復合材料。
上述的制備方法,優選的,所述s1步驟具體為:
s1-1、將納米硅和分散劑混合后,分散在溶劑中超聲得到納米硅分散液;
s1-2、將碳酸鋰和石墨混合后,分散在溶劑中得到碳酸鋰/石墨分散液;
s1-3、將所述納米硅分散液和碳酸鋰/石墨分散液混合后,烘干,在惰性氣氛下球磨得到碳酸鋰包覆硅/石墨混合物。
上述的制備方法,優選的,所述s1步驟具體為:
s1-1、將納米硅分散在溶劑中得到納米硅分散液;
s1-2、將碳酸鋰和石墨混合后,分散在溶劑中得到碳酸鋰/石墨分散液;
s1-3、將所述納米硅分散液和碳酸鋰/石墨分散液混合、球磨得到碳酸鋰/硅/石墨混合物。
上述的制備方法,優選的,所述s1步驟具體為:
s1-1、將納米硅和石墨混合,分散在溶劑中得到硅/石墨混合液;
s1-2、將碳酸鋰和增稠劑混合后,分散在溶劑中得到增稠劑/碳酸鋰混合液;
s1-3、將所述硅/石墨分混合液和增稠劑/碳酸鋰混合液混合得到碳酸鋰/硅/石墨混合液;
s1-4、將所述碳酸鋰/硅/石墨混合液烘干后球磨得到碳酸鋰/硅/石墨混合物。
上述的制備方法,優選的,所述增稠劑為瓊脂,所述瓊脂的添加量為0.5wt%。增稠劑可以增加碳酸鋰對硅/石墨的粘附性。
上述的制備方法,優選的,所述球磨過程中,球料比為50∶1~5∶1,球磨時間為1~10h球磨轉速為50~400r/min。
上述的制備方法,優選的,所述石墨、碳酸鋰的質量比為5~20∶1;所述石墨和納米硅的質量比為1∶10~100∶1;所述聚合物溶液的質量濃度為1%~50%。
上述的制備方法,優選的,所述石墨為鱗片石墨和/或石墨烯;所述聚合物溶液中的聚合物為瀝青、酚醛樹脂、葡萄糖、海藻酸鈉和聚丙烯酸酯類中一種或多種的混合物;所述溶劑為去離子水、nmp、無水乙醇、丙酮和四氫呋喃中的一種或者幾種的混合溶液。
作為一個總的技術構思,本發明還提供了一種上述硅碳復合材料或上述制備方法制備得到的硅碳復合材料在制備鋰離子電池中的應用。
上述的應用,優選的,所述應用方法為:將所述硅碳復合材料與導電碳黑、聚偏氟乙烯混合后,加入n-甲基吡咯烷酮調成漿狀物質;將所述漿狀物質涂在銅箔上,真空干燥后沖壓成鋰片;將所述鋰片與隔膜和電解液組裝成鋰離子電池。
本發明的有益效果:
(1)本發明提供了一種硅碳復合材料,以納米硅、石墨和碳酸鋰為原料,通過聚合物包覆后碳化得到。碳酸鋰是常用工業原料,也是電解液添加劑的一種,在惰性氣氛下碳酸鋰高溫加熱能釋放出二氧化碳造孔,為材料的體積變化預留空間,此外,碳酸鋰高溫還原為碳化鋰能起到補充鋰源、預嵌鋰的作用。以聚合物熱解后形成有機碳包覆在石墨、碳酸鋰、納米硅的表面,起到粘結納米硅與石墨的作用,同時碳包覆減少了球磨后材料的表面積,降低了副反應的發生,此外碳材料也提高了納米硅的導電率。而碳酸鋰的熱解增加了碳材料的孔隙率,能進一步緩沖硅的體積膨脹。因此,本發明的硅碳復合材料具有較高的導電性能兼具較為優異的循環穩定性。
(2)本發明提供了一種硅碳復合材料的制備方法,生產過程簡單,成分低廉、比較適合大規模的工業生產。在制備過程中,采用用高能球磨法將納米硅充分分散在石墨中,并使用聚合物包覆分散后的材料,緩沖硅材料在充放電過程中的體積變化。碳化處理后的硅碳復合材料具有良好的導電性。
(3)本發明提供了一種硅碳復合材料的制備方法,采用高能球磨處理納米硅和石墨,能減小材料粒徑,使硅與石墨接觸更加緊密。而采用液相分散-球磨處理的納米硅能較為均勻地分散在石墨材料中。
(4)本發明提供了一種硅碳復合材料的應用方法,硅碳復合材料可作為鋰離子電池負極材料,具有良好的循環性能和較高的容量、兼具較為優異的循環穩定性。
附圖說明
為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的一些實施例,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。
圖1為實施例1所制備得到的硅碳復合材料的首次充放電曲線。
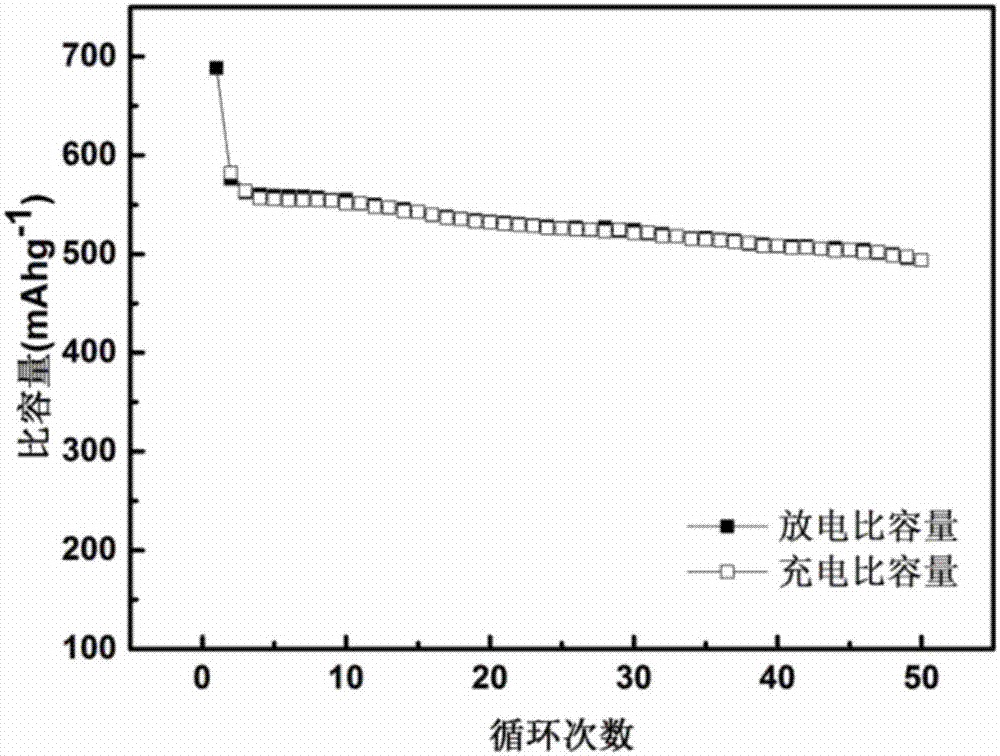
圖2為實施例1所制備得到的硅碳復合材料的循環性能。
具體實施方式
下面將結合實施例中的附圖,對本發明實施例中的技術方案進行清楚、完整地描述,顯然,所描述的實施例僅僅是本發明一部分實施例,而不是全部的實施例。基于本發明中的實施例,本領域普通技術人員所獲得的所有其他實施例,都屬于本發明保護的范圍。
實施例1
一種本發明的硅碳復合材料,其制備方法包括以下步驟:
s1、制備si@g材料:
1.1、稱取0.6g納米硅和0.1g分散劑ctab,分散在50ml的乙醇中超聲半小時得到納米硅分散液;
1.2、稱取0.3g碳酸鋰和3g鱗片石墨,加入60ml的乙醇中攪拌2h得到碳酸鋰/石墨分散液;
1.3、將步驟1.1的納米硅分散液滴加入步驟1.2的碳酸鋰/石墨分散液分散液中,攪拌2h后超聲處理40min,(超聲波細胞破碎儀工作頻率為22±1khz)得到均一的分散液;
1.4、將步驟1.3中均一的分散液真空抽濾得到濾渣,將濾渣在真空干燥箱內以80℃烘干;
1.5、將步驟1.4得到的的烘干產物放入球磨罐中,在氬氣氛圍下,以球料比30∶1、轉速350r/min球磨1h,球磨結束后把材料過400目篩,得到碳酸鋰包覆硅/石墨混合物(si@g材料)。
s2、制備前驅體材料:
2.1、取1g瀝青,均勻分散在100ml乙醇中得到瀝青分散液;
2.2、將步驟s1的si@g材料加入到步驟2.1的瀝青分散液中充分攪拌,分散均勻后在80℃油浴攪拌蒸干,然后在真空干燥箱內以80℃烘干,得到前驅體材料。
s3、制備si@g-c材料:
將步驟s2中的前驅體置于通氬氣的管式爐中,以5℃/min的升溫速率從室溫升到200℃,在200℃下保溫2h,然后繼續升溫到900℃,并在900℃下保溫6h,然后隨爐冷卻,得到si@g-c,即硅碳復合材料。
將實施例1的si@g-c與導電碳黑、聚偏氟乙烯(pvdf)以質量比8∶1∶1混合得到混合物。將混合物研磨,加入適量n-甲基吡咯烷酮(nmp)調成漿狀物質(nmp作為pvdf溶劑調和混合物,其混合物與nmp的質量體積比為:2g∶0.4~0.5ml(滴管8~10滴));將漿狀物質涂在銅箔上,再于真空干燥箱中120℃干燥4h,然后沖壓成圓形鋰片。將鋰片、隔膜、電解液(電解液的成份為1mol/llipf6inec/dmc(1∶4)+2%fec)組裝成鋰離子扣式電池。
檢測鋰離子電池充放電性能:
首先以50mag-1的電流密度充放電1次來進行活化,再以100mag-1的電流密度對該電池進行充放電測試,得到的首次充放電曲線如圖1。從圖1中可知:實施例1的鋰離子電池的首次放電比容量為688.25mahg-1,首次充電比容量為582.00mahg-1,首次庫倫效率達到84.56%。圖2為鋰離子電池的循環性能,從圖2中可知:經50次循環后,放電比容量仍有493.50mahg-1,容量保持率為85.67%。
實施例2
一種本發明的硅碳復合材料,其制備方法包括以下步驟:
s1、制備硅-石墨碳酸鋰混合液:
1.1、稱取0.6g納米硅分散在50ml的去離子水中得到納米硅分散液;
1.2、稱取0.3g碳酸鋰和3g鱗片石墨,加入100ml的去離子水中分散得到碳酸鋰/石墨分散液;
1.3、將步驟1.1的納米硅分散液滴加入步驟1.2的碳酸鋰/石墨分散液中,以350r/min轉速球磨1h得到碳酸鋰/硅/石墨混合物。
s2、制備前驅體材料:
2.1、取1.5g葡萄糖溶于250ml去離子水中得到葡萄糖溶液;
2.2、將步驟s1的碳酸鋰/硅/石墨混合物逐滴加入到步驟2.1的葡萄糖溶液中充分攪拌,超聲分散2h(超聲波細胞破碎儀工作頻率為22±1khz)得到懸浮液,再將懸浮液以15ml/min的速度在噴霧干燥器中噴霧干燥,入口溫度和出口溫度分別保持在160℃和110℃,得到前驅體材料。
s3、制備si@g-c材料:
將步驟s2中的前驅體置于通氬氣的管式爐中,以5℃/min的升溫速率從室溫升到200℃,在200℃下保溫2h,然后繼續升溫到800℃,并在800℃下保溫6h,然后隨爐冷卻,得到si@g-c,即硅碳復合材料。
將實施例2的si@g-c與導電碳黑、聚偏氟乙烯(pvdf)以質量比8∶1∶1混合得到混合物。將混合物研磨,加入適量n-甲基吡咯烷酮(nmp)調成漿狀物質(nmp作為pvdf溶劑調和混合物,其混合物與nmp的質量體積比為:2g∶0.4~0.5ml(滴管8~10滴));將漿狀物質涂在銅箔上,再于真空干燥箱中120℃干燥4h,然后沖壓成圓形鋰片。將鋰片、隔膜、電解液組裝成鋰離子扣式電池。
檢測鋰離子電池充放電性能:
首先以50mag-1的電流密度充放電1次來進行活化,再以100mag-1的電流密度對該電池進行充放電測試。電池的首次放電比容量為602.26mahg-1,首次充電比容量為515.96mahg-1,首次庫倫效率達到83.79%。經50次循環后,放電比容量仍有417.29mahg-1,容量保持率為82.75%。
實施例3
一種本發明的硅碳復合材料,其制備方法包括以下步驟:
s1、制備硅-石墨碳酸鋰混合液:
1.1、稱取0.6g納米硅和3g鱗片石墨混合,分散在100ml的乙醇得到硅/石墨混合液;
1.2、稱取0.2g碳酸鋰和0.1g瓊脂,加入80ml的去離子水中得到瓊脂/碳酸鋰混合液;
1.3、將步驟1.2的瓊脂/碳酸鋰混合液滴加入步驟1.1的硅/石墨混合液中,不斷攪拌,超聲處理2h(超聲波細胞破碎儀工作頻率為22±1khz)得到碳酸鋰/硅/石墨混合液;
1.4、將碳酸鋰/硅/石墨混合液在80℃油浴攪拌蒸干,將蒸干后的干燥物放入球磨罐中,氬氣氛圍下,以球料比30∶1、轉速350r/min,球磨2h,球磨結束后把材料過400目篩,得到碳酸鋰/硅/石墨混合物,即si@g材料。
s2、制備前驅體材料:
2.1、取1.5g一水合檸檬酸溶于100ml體積分數為50%的乙醇溶液中得到檸檬酸溶液;
2.2、將步驟s1的si@g材料分散到步驟2.1的檸檬酸溶液中攪拌3h,然后在80℃的油浴鍋中蒸干,得到前驅體材料。
s3、制備si@g-c材料:
將步驟s2中的前驅體置于通氬氣的管式爐中,以5℃/min的升溫速率從室溫升到800℃,在800℃下保溫6h,然后隨爐冷卻,得到si@g-c,即硅碳復合材料。
將實施例3的si@g-c與導電碳黑、聚偏氟乙烯(pvdf)以質量比8:1:1混合得到混合物。將混合物研磨,加入n-甲基吡咯烷酮(nmp)調成漿狀物質(nmp作為pvdf溶劑調和混合物,其混合物與nmp的質量體積比為:2g∶0.4~0.5ml(滴管8~10滴));將漿狀物質涂在銅箔上,再于真空干燥箱中120℃干燥4h,然后沖壓成圓形鋰片。將鋰片、隔膜、電解液組裝成鋰離子扣式電池。
檢測鋰離子電池充放電性能:
首先以50mag-1的電流密度充放電1次來進行活化,再以100mag-1的電流密度對該電池進行充放電測試。電池的首次放電比容量為682.23mahg-1,首次充電比容量為525.17mahg-1,首次庫倫效率達到76.98%。經50次循環后,放電比容量為329.21mahg-1,容量保持率為62.68%。
綜合上述實施例1、2、3,分別采用了不同的碳源和干燥方式,有對比可知采用瀝青作為碳源合成的材料循環性能最優,容量損失最小。干燥方式對材料性能的影響較小。
以上所述僅為本發明的較佳實施例而已,并不用以限制本發明,凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!