一種N?Fe2O3/氮硫雙摻雜石墨烯復合電極材料及制備方法與流程
本發明涉及一種復合材料的制備方法,尤其涉及一種以氮硫雙摻雜石墨烯為基體定域負載氮摻雜三氧化二鐵納米粒子復合材料的制備方法,所制備的復合材料可應用于超級電容器領域。
背景技術:
隨著環境污染和能源短缺問題日益嚴重,尋找新型可替代能源和儲能裝置已經迫在眉睫。超級電容器由于比電容高,循環穩定性好,工作溫度范圍寬,綠色環保,充電效率高和充電時間短等優點而引起了廣泛關注。石墨烯具有單層二維結構,由于其較大的比表面積、超高的穩定性和優異的電導率等性能已成為電化學領域的研究熱點。然而文獻表明,石墨烯的實際容量小于理論容量,且衰減較快,這需要進行雜原子的摻雜調節石墨烯的電子結構。(chenw,shij,zhut等.electrochimicaacta,2015,177:327-334;yiht,zhuyq,chenxy等journalofalloys&compounds,2015,649:851-858.)在石墨烯片層間引入n、s等雜原子可以改進石墨烯的電子電導率和倍率性能,n的摻雜可以在石墨烯晶格結構中引入更多的缺陷,并活化與n相連的c原子,而s的引入能夠有效降低反應所需的活化能,提高石墨烯的局域反應活性。此外,由于石墨烯密度和自旋密度的重新分布,雙摻雜可以產生更多缺陷孔,在與金屬氧化物發生氧化還原反應存儲電荷的過程中產生顯著的協同效應。
簡單過渡金屬及過渡金屬氧化物因為具有較高的理論電容量,因此受到廣泛的關注,其中鐵的氧化物具有較高的理論比容量、廉價易得和環境友好等優點,受到了較多的研究。但循環穩定性較差,阻礙了實際電極材料的進一步發展。氮(n)被認為是最有可能實現n型fe2o3的摻雜元素,因為它的離子半徑與氧(o)接近,而且n-fe2o3具有更多的活性位點,有利于電解質離子的傳輸,從而提高電化學性能。將n-fe2o3與氮硫雙摻雜石墨烯材料復合能有效避免石墨烯間堆疊,使n-fe2o3的高電容與改性石墨烯材料的大比表面積優勢相互結合,達到雙電層電容和贗電容的完美結合,極大的改良了復合材料的電容性能。目前,大量的文獻進行摻雜都是通過兩步或以上方法,先分別合成納米粒子和基體材料,再進行納米粒子和改性基體材料的復合。(wangm,huangy,chenx等.journalofalloys&compounds,2017,691:407-415.)公開號為cn105826572a的中國專利文獻公開了一種n,s雙摻雜碳納米管包覆fexc催化劑的制備方法,先制得催化劑前驅體,再經過高溫煅燒和酸刻蝕得到催化劑材料。
本發明首次采用一步水熱法合成了n-氧化物負載雙摻雜石墨烯復合材料,并將氮摻雜氧化物首次應用到超級電容器的電極材料上,以常用的九水合硝酸鐵,氧化石墨烯,乙醇,乙二醇和硫氰酸銨為原料,通過簡便的一步水熱法合成n-fe2o3/氮硫雙摻雜石墨烯復合材料,實驗操作簡單,成本低廉,所制備的n-fe2o3能大量的儲存電荷,雙摻雜石墨烯則能提供電子傳輸的通道和結構穩定骨架,有利于電解質離子的自由遷移,從而充分發揮n-fe2o3納米粒子和石墨烯之間的協同效應使得雙電層電容和贗電容的完美結合,得到兼具高功率密度和高能量密度的超級電容器材料。
技術實現要素:
本發明的目的在于:采用簡便的一步水熱法制備出一種n-fe2o3/氮硫雙摻雜石墨烯復合電極材料。
本發明是通過如下技術方案實現的:
一種n-fe2o3/氮硫雙摻雜石墨烯復合電極材料,所述電極材料是由n-fe2o3和氮硫雙摻雜石墨烯復合而成的,所述n-fe2o3負載于所述氮硫雙摻雜石墨烯表面;所述n-fe2o3中,n元素摻雜在fe2o3晶格表面;所述氮硫雙摻雜石墨烯中,n、s元素均摻雜在石墨烯中;在所述n-fe2o3/氮硫雙摻雜石墨烯復合電極材料中,n的摻雜量為2.30wt%~2.39wt%,s的摻雜量為0.70%~0.90wt%。
一種n-fe2o3/氮硫雙摻雜石墨烯復合電極材料的制備方法,步驟如下:
步驟1、將氧化石墨烯置于乙醇和乙二醇的混合溶液中超聲分散,得到穩定的均相氧化石墨烯的分散溶液a;
步驟2、將九水合硝酸鐵和氮硫源加入到步驟1的分散溶液a中,攪拌均勻得到混合溶液b;
步驟3、將步驟2的混合溶液b轉移到聚四氟乙烯水熱釜中進行恒溫水熱反應,反應完畢后得到混合液c;
步驟4、將步驟3的混合液c抽濾、洗滌、干燥,得到產物n-fe2o3/氮硫雙摻雜石墨烯復合材料。
步驟1中,所述分散溶液a中,所述氧化石墨烯與乙醇和乙二醇的混合溶液的用量比為9mg:7ml,所述乙醇和乙二醇的混合溶液中,乙醇的體積和乙二醇的體積比為(0.5~2):1。
進一步,乙醇和乙二醇的體積比為2:1。
步驟2中,所述混合溶液b中,所使用的九水合硝酸鐵和氮硫源與所述分散溶液a的用量比為0.004mol:0.012~0.028mol:70ml。
進一步,所述氮硫源為硫氰酸銨或過硫酸銨。
進一步,所述氮硫源為硫氰酸銨,所述九水合硝酸鐵和硫氰酸銨的摩爾比為1:6。
步驟3中,所述恒溫水熱反應的溫度為160~200℃,反應時間為18~30h。
進一步,恒溫熱反應的溫度為180℃,反應時間為24h。
本發明與現有技術相比具有以下優點:
1、乙醇和乙二醇的混合溶液使納米粒子粒徑更小,分散更均勻,高溫作用下硫氰酸銨分解釋放出s2-和nh4+,將n和s摻雜到片狀石墨烯結構中,形成雙摻雜結構,nh3不僅作為堿源,還能夠提供n形成n-fe2o3,石墨烯和fe2o3晶格表面氮的共同作用使fe2o3能夠更均勻穩定分散在雙摻雜石墨烯表面,達到雙電層電容和贗電容的完美結合,提高電化學循環穩定性。
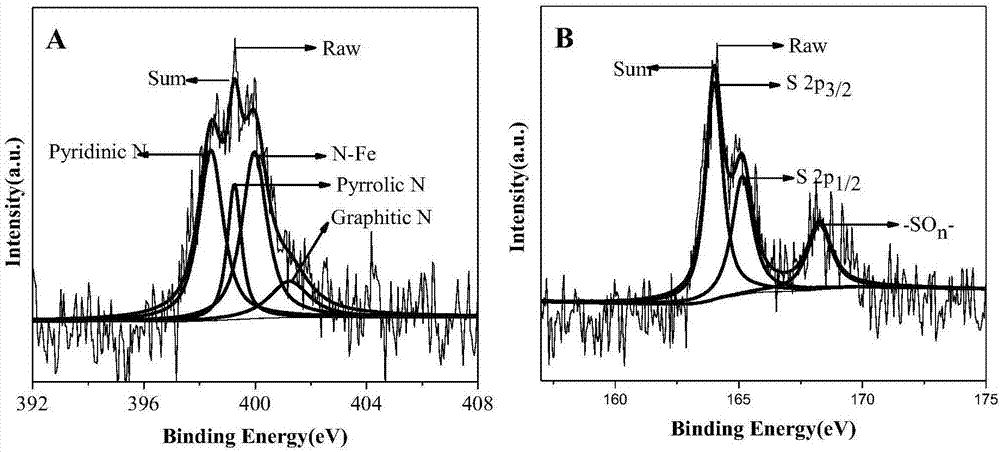
2、xps表明采用本發明方法制備的n-fe2o3/氮硫雙摻雜石墨烯復合材料中n成功地被摻雜到fe2o3晶格表面和石墨烯中,同時s被摻雜到石墨烯中。n以碳氮鍵、吡啶氮、吡咯氮和n-fe特征峰形式存在,s以s2p3/2、s2p1/2和-son-形式存在。其中n的摻雜量為2.30wt%~2.39wt%,s的摻雜量為0.70%~0.90wt%。
3、本發明首次用一步水熱法合成納米粒子合成n-fe2o3納米粒子負載雙摻雜石墨烯材料,原料簡單,綠色環保,合成過程簡單無污染。
4、將所制備的材料首次應用于超級電容器的電極材料,在1.0a/g的恒電流密度下,其單極比電容可達1263.7fg-1。
附圖說明
圖1為所制備n-fe2o3/三維氮硫雙摻雜石墨烯及n-fe2o3單獨組分的x射線衍射譜圖。
圖2為n-fe2o3/氮硫雙摻雜石墨烯的x射線光電子能譜分析,其中,圖a為n1s能譜峰,圖b為s2p能譜峰。
圖3為不同放大尺度下n-fe2o3/氮硫雙摻雜石墨烯的透射電鏡照片。
圖4為n-fe2o3,氮硫雙摻雜石墨烯,n-fe2o3/氮硫雙摻雜石墨烯在1ag-1條件下的恒電流充放電曲線圖。
圖5為n-fe2o3/氮硫雙摻雜石墨烯在不同電流密度下的恒電流充放電曲線圖。
具體實施方式
下面結合具體實施實例對本發明做進一步說明。
實施例1
稱取1.6159gfe(no3)3·9h2o(0.004mol)、0.9134gnh4scn(0.012mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為2:1)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,180℃反應24h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,研磨得到n-fe2o3納米粒子。樣品在2θ為24.15°、33.16°、35.63°、40.82°、49.46°、54.07°、57.60°、62.44°、64.00°、71.95°和75.46°均出現了x射線的衍射峰,分別可以與fe2o3晶體的(012)、(104)、(110)、(113)、(024)、(116)、(018)、(214)、(300)、(1010)和(220)晶面與標準譜相對應,如圖1,圖中衍射峰均為石墨烯和三氧化二鐵的特征衍射峰,表明生成了fe2o3納米粒子。將上述制備的n-fe2o3納米材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了682.4fg-1;經過2000次充放電測試之后比容量仍保持在78%以上。
實施例2
稱取90mg氧化石墨烯、0.9134gnh4scn(0.012mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為2:1)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,180℃反應24h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,即得氮硫雙摻雜石墨烯。氮硫雙摻雜石墨烯具有較大的比表面,氮和硫的摻雜激活了與其相鄰的碳原子,增加了活性位點的數量,有利于電解質離子與活性物質發生反應。將上述制備的氮硫雙摻雜石墨烯材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了643.2fg-1;經過2000次充放電測試之后比容量仍保持在93%以上。
實例3
稱取90mg氧化石墨烯、1.6159gfe(no3)3·9h2o(0.004mol)、1.8269gnh4scn(0.024mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為0.5:1)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,180℃反應24h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,即得n-fe2o3/三維氮硫雙摻雜石墨烯。產品經x射線光電子能譜分析(xps)氮以石墨氮、吡啶氮、吡咯氮和n-fe形式存在,硫以s2p3/2,s2p1/2and-son-形式存在,表明氮原子成功地摻雜到fe2o3的晶格表面,氮、磷原子成功地摻雜到石墨烯的晶格結構中,如圖2。三維氮硫雙摻雜石墨烯具有較大的比表面,氮和硫的摻雜激活了與其相鄰的碳原子,增加了活性位點的數量,形成了更多的缺陷孔,有利于電解質離子與活性物質發生反應。將上述制備的n-fe2o3/氮硫雙摻雜石墨烯材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了1263.7fg-1,如圖4。在10ag-1大的電流密度下,電容仍然有在983fg-1,如圖5,放電曲線存在平緩的放電平臺和明顯的氧化還原反應贗電容放電過程,進一步說明該復合材料用作電容器的電極材料在大電流密度下有好的穩定性。經過2000次充放電測試之后比容量仍保持在87%以上。
實施例4
稱取90mg氧化石墨烯、1.6159gfe(no3)3·9h2o(0.004mol)、1.5224gnh4scn(0.020mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為2:1)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,160℃反應30h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,即得n-fe2o3/氮硫雙摻雜石墨烯。將上述制備的n-fe2o3/氮硫雙摻雜石墨烯材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了364.8fg-1;經過2000次充放電測試之后比容量仍保持在53%以上。
實例5
稱取90mg氧化石墨烯、1.6159gfe(no3)3·9h2o(0.004mol)、2.1314gnh4scn(0.028mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為1:1)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,160℃反應18h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,即得n-fe2o3/氮硫雙摻雜石墨烯。將上述制備的n-fe2o3/氮硫雙摻雜石墨烯材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了268.6fg-1;經過2000次充放電測試之后比容量仍保持在62%以上。
實例6
稱取90mg氧化石墨烯、1.6159gfe(no3)3·9h2o(0.004mol)、2.352g(nh4)2s2o8(0.012mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為1:1)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,180℃反應24h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,即得n-fe2o3/氮硫雙摻雜石墨烯。如圖3所示,n-fe2o3納米粒子均勻分散在氮硫雙摻雜石墨烯表面,粒徑范圍在15-25nm。將上述制備的n-fe2o3/氮硫雙摻雜石墨烯材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了968.6fg-1;經過2000次充放電測試之后比容量仍保持在78%以上。
實例7
稱取90mg氧化石墨烯、1.6159gfe(no3)3·9h2o(0.004mol)、4.704g(nh4)2s2o8((0.024mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為0.5:1)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,200℃反應16h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,即得n-fe2o3/氮硫雙摻雜石墨烯。將上述制備的n-fe2o3/氮硫雙摻雜石墨烯材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了842.6fg-1;經過2000次充放電測試之后比容量仍保持在72%以上。
實例8
稱取90mg氧化石墨烯、1.6159gfe(no3)3·9h2o(0.004mol)、2.352g(nh4)2s2o8(0.012mol)超聲分散在70ml無水乙醇和乙二醇的混合溶液(體積比為1:2)中,超聲分散1h,將混合溶液轉移到100ml水熱釜中,200℃反應30h。反應完成后自然冷卻至室溫,無水乙醇和去離子水交替離心洗滌十次,然后在真空干燥箱中于60℃干燥12小時,即得n-fe2o3/氮硫雙摻雜石墨烯。將上述制備的n-fe2o3/氮硫雙摻雜石墨烯材料進行充放電實驗,電流密度為1ag-1時,比容量值達到了768.4fg-1;經過2000次充放電測試之后比容量仍保持在78%以上。
以上所揭露的僅為本發明的優選實施例而已,當然不能以此來限定本發明的權利范圍,因此,以本發明范圍內所做的等同變化,仍屬于本發明所涵蓋的范圍。本發明所屬技術領域的技術人員可以對所描述的具體實施例做各種修改或補充或采用類似的方式替代,但并不會偏離本發明的精神或者超越所附權利要求書所定義的范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!