一種凝膠電解質的制備方法及由其制備的凝膠電解質與流程
本發明涉及鋰離子電池領域,且特別涉及一種凝膠電解質的制備方法及由其制備的凝膠電解質。
背景技術:
目前,鋰離子電池主要應用隔膜/電解液體系。其中,隔膜主要是聚烯烴微孔膜,例如聚丙烯微孔膜(Polypropylene,簡稱PP)、聚乙烯微孔膜(Polyethylene,簡稱PE)和PP-PE多層復合隔膜等。但是,聚烯烴隔膜存在熱穩定性不好、高溫尺寸穩定性很差和對電解液浸潤性差的缺點,同時,鋰離子電池在使用時需要引入大量的可燃性有機電解液。由于上述問題的存在,當鋰離子電池在過充和過熱等非正常環境下使用時都極易引發安全事故。解決上述問題一個有效的途徑是制備性能優異的聚合物電解質,取代鋰離子電池應用的隔膜/電解液體系。
現有的凝膠聚合物電解質最常用的聚合物骨架材料有聚氧化乙烯(Homopolymer,簡稱PEO)、聚甲基丙烯甲酯(Polymethylmethacrylate,簡稱PMMA)、聚丙烯腈(Polyacrylonitrile,簡稱PAN)、聚偏氟乙烯(Polyvinylidene fluoride,簡稱PVDF)和聚偏氟乙烯-六氟丙烯共聚物共聚物(Poly vinylidenefluoride-hexafluoro propylene,簡稱PVDF-HFP)等。但是,上述任意一種骨架材料均存在一定的缺點,例如:PEO、PVDF和PVDF-HFP存在結晶的問題,PMMA是存在機械強度差的問題,PAN存在與鋰金屬相容性差的問題,由于存在這些缺點導致單一組分的骨架材料無法完全滿足鋰離子電池的使用要求。此外,即使將上述骨架材料與其他聚合物進行共混雖然能解決以上問題,但是,簡單共混時,其中的某些組分極易被有機電解液中的極性溶劑溶脹,產生制備的凝膠電解質的熱穩定性和尺寸穩定性差的問題。
技術實現要素:
本發明的目的在于提供一種凝膠電解質的制備方法,以解決凝膠電解質的熱穩定性差和尺寸穩定性差的問題。
本發明的另一目的在于提供一種凝膠電解質,通過上述的制備方法制得,具有較好的熱穩定性和尺寸穩定性。
本發明解決其技術問題是采用以下技術方案來實現的。
本發明提出一種凝膠電解質的制備方法,包括:以自由基聚合反應得到PS-PEO-PS。將PS-PEO-PS與骨架材料共混得到共混多孔膜。將共混多孔膜在催化劑和交聯劑存在環境下發生傅克反應得到交聯多孔膜。交聯多孔膜吸收電解液后制得凝膠電解質。
一種凝膠電解質,根據上述的凝膠電解質的制備方法制得。
本發明較佳實施例的凝膠電解質的制作方法,通過將共混多孔膜交聯得到交聯多孔膜,得到熱穩定性和尺寸穩定性好的凝膠電解質。本制備方法是一種靈活高效的交聯方法,交聯度可以方便地通過反應底物和反應條件得到調控,以滿足不同的實際需求。
本發明較佳實施例的凝膠電解質具有較好的熱穩定性。
附圖說明
為了更清楚地說明本發明實施例的技術方案,下面將對實施例中所需要使用的附圖作簡單地介紹,應當理解,以下附圖僅示出了本發明的某些實施例,因此不應被看作是對范圍的限定,對于本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他相關的附圖。
圖1為本發明各實施例獲得樣品的DSC分析曲線;
圖2為本發明各實施例獲得樣品的TGA分析曲線;
圖3為本發明各實施例獲得樣品的高溫下孔隙率分析圖;
圖4為本發明各實施例獲得樣品的熱收縮性分析圖;
圖5為本發明各實施例獲得樣品的交流阻抗譜;
圖6為本發明各實施例獲得樣品的線性伏安掃描曲線;
圖7為本發明各實施例獲得樣品的容量保留率曲線。
具體實施方式
為使本發明實施例的目的、技術方案和優點更加清楚,下面將對本發明實施例中的技術方案進行清楚、完整地描述。實施例中未注明具體條件者,按照常規條件或制造商建議的條件進行。所用試劑或儀器未注明生產廠商者,均為可以通過市售購買獲得的常規產品。
下面對本發明實施例的凝膠電解質的制備方法及由其制備的凝膠電解質進行具體說明。
一種凝膠電解質的制備方法,其反應式可以如下式所示。
可以理解,凝膠電解質的制備方法主要包括:
以活性自由基聚合反應得到PS-PEO-PS。
例如:以聚氧化乙烯(PEO)和2-溴異丁酰溴(2-Bromoisobutyryl bromide,簡稱BIBB)為底物,以二氯甲烷(Dichloromethane,簡稱DCM)為第一溶劑,并將二氯甲烷分為第一部分二氯甲烷和第二部分二氯甲烷,以三乙胺(Triethylamine,簡稱TEA)為縛酸劑,反應制得Br-PEO-Br。
具體地,將聚氧化乙烯溶于第一部分二氯甲烷后,向混合液中加入三乙胺。將2-溴異丁酰溴溶于第二部分二氯甲烷后。在無氧無水環境下,將上述的兩種溶液混合后,在25~35℃的溫度下攪拌10~18h得到第一中間體。依次除去第一中間體的三乙胺溴酸鹽、過量的2-溴異丁酰溴和二氯甲烷后,干燥制得Br-PEO-Br。制得的Br-PEO-Br為用于制備PS-PEO-PS的大分子引發劑。
除去三乙胺溴酸鹽和過量的2-溴異丁酰溴可以采用如下方法:將第一中間體濾去三乙胺溴酸鹽得到濾液,再依次使用飽和碳酸氫鈉(NaHCO3)溶液和去離子水分別洗滌多次后,將洗滌后的反應產物加入到適量的無水硫酸鈉(Na2SO4)中進行除水,將吸水后的硫酸鈉濾去得到濾液。通過上述操作,實現將反應產物中的三乙胺溴酸鹽和過量的2-溴異丁酰溴除去并得到干燥的反應產物。
除去DCM可以采用如下方法:將上述的濾液進行蒸發后,使用乙醚進行沉降得到固體,用乙醚洗滌固體多次后,在25~40℃的烘箱中烘干白色固體直至恒量,白色固體即為Br-PEO-Br。作為優選,烘干時間以20~30h為宜。
以Br-PEO-Br、1,4-二氧六環和苯乙烯單體為底物,以溴化亞銅為催化劑,以N,N,N',N,'N”-五甲基二亞乙基三胺(N,N,N',N,'N”-Pentamethyldiethylenetriamine,簡稱PMDETA)為催化劑配體,反應制得PS-PEO-PS。具體地,將Br-PEO-Br與溴化亞銅(CuBr)混合成反應體系后,在無氧無水的環境下,依次向其中加入1,4-二氧六環、苯乙烯單體和N,N,N',N,'N”-五甲基二亞乙基三胺并在100~120℃的溫度下攪拌12~36h得到第二中間體。除去第二中間體中的銅鹽后沉降干燥得到白色固體,白色固體即為PS-PEO-PS三嵌段共聚物。
除去第二中間體中的銅鹽可以采用如下方法:將第二中間體暴露于空氣中冷卻,冷卻溫度以20~25℃為宜。向第二中間體加入四氫呋喃(Tetrahydrofuran,簡稱THF)稀釋后,過柱除去銅鹽。其中,柱子可以選用堿性三氧化二鋁柱,也可以選用硅膠柱。作為優選,柱子選用堿性三氧化二鋁柱。過柱后,將得到的稀釋液經行濃縮后加入到正己烷中析出、過濾得到濾渣。濃縮例如可以在旋轉蒸發儀中進行,將稀釋液加入到旋轉蒸發儀并將稀釋液旋至少量。將濾渣在30~40℃的烘箱中烘干直至得到白色絮狀固體,白色絮狀固體即為PS-PEO-PS三嵌段共聚物。
承上述,通過對反應底物的選擇和反應條件的控制,實現了PS-PEO-PS三嵌段共聚物制備的可控性,制備得到分子量分布較窄的三嵌段共聚物。
將PS-PEO-PS與骨架材料共混得到共混多孔膜。共混多孔膜可以采用以下方法制得:
以PS-PEO-PS和骨架材料通過相轉化法制得共混多孔膜。將PS-PEO-PS和骨架材料混合后,向其中加入第二溶劑并在75~85℃的溫度下攪拌直至PS-PEO-PS和骨架材料形成均一穩定的溶液。其中,第二溶劑選自N,N-甲基吡咯烷酮(1-Methyl-2-pyrrolidinone,簡稱NMP)。向均一穩定的溶液中加入成孔劑并在75~85℃的溫度下攪拌20~30h后得到鑄膜液。成孔劑選自甘油(Glycerine,簡稱Gl)。將鑄膜液冷卻至20~25℃的溫度后進行脫泡。脫泡可以采用真空脫泡、超聲脫泡和靜置脫泡中的一種脫泡方式進行,只要將鑄膜液中的氣泡脫掉即可。可以理解,鑄膜液在澆鑄之前進行脫泡處理,以保證鑄膜液的澆鑄效果。
將脫泡后的鑄膜液澆鑄于培養皿中并在80~100℃的溫度下進行第一次烘干,得到固態膜,第一次烘干的時間以3~5h為宜。之后,在120~140℃的溫度下對經過第一次烘干的固態膜進行第二次烘干,第二次烘干的時間以12~36h為宜,得到共混多孔膜。
通過第一次烘干,除去鑄膜液中的第二溶劑;通過第二次烘干,除去鑄膜液中的成孔劑,得到無雜質的共混多孔膜。此外,在第一次烘干和第二次烘干的過程中,不一定要限制烘干的溫度和烘干的時間,只要保證將鑄膜液中的N,N-甲基吡咯烷酮和甘油除去即可。
承上述,骨架材料選自聚氧化乙烯(PEO)、聚甲基丙烯甲酯(PMMA)、聚丙烯腈(PAN)、聚偏氟乙烯(PVDF)以及聚偏氟乙烯-六氟丙烯共聚物(PVDF-HFP)中的至少一種。
作為優選,骨架材料選自PVDF。可以理解,例如以PVDF為骨架材料制備共混多孔膜時,則制備的共混多孔膜為PVDF/PS-PEO-PS共混多孔膜。
將共混多孔膜在催化劑和交聯劑存在下發生傅克反應,得到交聯多孔膜。在無氧無水的環境下,以共混多孔膜為底物,以三氯化鐵(FeCl3)為催化劑,以1,2-二氯乙烷為溶劑,以二甲氧基甲烷(FDA)和乙二醇二甲醚中的至少一種為交聯劑,使得共混多孔膜發生傅克反應得到交聯多孔膜。
具體地,將三氯化鐵和二甲氧基甲烷加入1,2-二氯乙烷中形成反應體系,并將反應體系在0~10℃的溫度下攪拌均勻。向反應體系內加入共混多孔膜,在25~80℃的溫度下經反應3~36h得到交聯多孔膜。可以理解,通過對共混膜中PS-PEO-PS中的苯環之間的交聯,實現同一片膜內部的嵌段共聚物之間的交聯從而得到的超交聯結構,即為交聯多孔膜,也可以理解,上述反應過程即為傅克反應。
為了保證交聯多孔膜的使用效果,避免交聯多孔膜的老化或污染,在交聯多孔膜使用前需要將其制備過程帶有的催化劑除去,即將交聯多孔膜中含有的三氯化鐵除去。將制得的交聯多孔膜用丙酮洗滌多次后,用1mol/L的稀鹽酸溶液對其洗滌多次,再用去離子水對其洗滌多次后,用甲醇對其洗滌多次。
使用甲醇對交聯多孔膜的洗滌可以采用如下方法:將去離子水洗滌多次后的交聯多孔膜加入到索氏提取器中,使用甲醇對其提取多次直至將其中殘留的三氯化鐵徹底除去。通過上述操作,為了保證將交聯多孔膜中的三氯化鐵無殘留,也可以理解,得到交聯多孔膜即為熱穩定性好且不溶脹的交聯多孔膜。
將制備的交聯多孔膜置于電解液中,充分吸收電解液后制得凝膠電解質。可以理解,制備的交聯多孔膜即為交聯凝膠聚合物電解質(Cross-linked Gel Polymer Electrolytes),簡稱CPE。通過交聯,將共混后的共混多孔膜制得交聯多孔膜,并使得制備的交聯多孔膜具有較好的熱穩定性和尺寸穩定性。本交聯方法是一種靈活高效的交聯方法,交聯度可以方便地通過反應底物和反應條件得到調控,以滿足不同的實際需求。
根據上述的凝膠電解質的制備方法制得的凝膠電解質。是一種離子電導率高、熱穩定性好和尺寸穩定性高的電解質。
綜上所述,本發明實施例的凝膠電解質的制作方法,通過將共混多孔膜交聯得到交聯多孔膜,得到熱穩定性好和尺寸穩定性好的凝膠電解質。本制備方法是一種靈活高效的交聯方法,交聯度可以方便地通過反應底物和反應條件得到調控,以滿足不同的實際需求。
本發明實施例的凝膠電解質具有較好的熱穩定性和尺寸穩定性。
以下結合實施例對本發明的特征和性能作進一步的詳細描述。
實施例1
以PS15-PEO-PS15三段共聚物制備交聯多孔膜。
a)在無水無氧的環境下,將150ml的二氯甲烷和8.8g的聚氧化乙烯(Mn=8000g/mol)混合均勻后,向其中加入4.46ml的三乙胺并再混合均勻,得到第一混合液。
在無水無氧的環境下,將10mL的二氯甲烷和4.14mL的2-溴異丁酰溴混合均勻,得到第二混合液。
在冰水浴的溫度下,將第二混合液加入到第一混合液后得到反應體系,將反應體系在28℃的溫度下攪拌18h,得到第一中間體。使用飽和碳酸氫鈉溶液對第一中間體洗滌三次,再使用去離子水對第一中間體洗滌三次。向洗滌后的第一中間體內加入適量無水硫酸鈉進行干燥,過濾,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到冷的無水乙醚中沉降析出,過濾,得濾渣。用乙醚對濾渣洗滌多次后在30℃的溫度下烘干25h,得到Br-PEO-Br。
在無水無氧的環境下,將2g的Br-PEO-Br和0.072g的溴化亞銅混合均勻后,向其中加入20mL的1,4-二氧六環和1.15mL的苯乙烯單體混合均勻,得到反應體系。向反應體系內加入0.156mL的N,N,N',N,'N”-五甲基二亞乙基三胺并在115℃的溫度下反應24h,得到第二中間體。將第二中間體暴露在空氣中待冷卻至室溫后,向反應產物中加入四氫呋喃進行稀釋,得到稀釋液。將稀釋液過堿性三氧化二鋁短柱除去銅鹽,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到正己烷中沉降析出,過濾,得濾渣。將濾渣在40℃的溫度下烘干得到PS15-PEO-PS15三段共聚物。
b)將4g的聚偏氟乙烯(PVDF)和1g的PS15-PEO-PS15混合均勻后,向其中加入35.1mL的N,N-甲基吡咯烷酮,并在80℃的溫度下攪拌使形成均一的溶液后,向其中加入3.51mL的甘油,繼續在80℃的溫度下攪拌24h后,得到反應產物。將反應產物冷卻到室溫后脫泡。將脫泡后的反應產物澆鑄到培養皿中,在90℃的溫度下烘干4h后,再在溫度為120℃的真空環境中烘干24h,得到PVDF/PS15-PEO-PS15共混多孔膜。
c)在無水無氧的環境下,向4.875g的無水三氯化鐵中依次加入20mL的1,2-二氯乙烷和2.65mL的二甲氧基甲烷,得到反應體系。
在冰水浴的溫度下,將反映體系充分攪拌。向反應體系中加入500mg的PVDF/PS15-PEO-PS15共混多孔膜,在80℃的溫度下反應26h得到反應產物。將反應產物依次用丙酮洗滌3次、1mol/L的稀鹽酸溶液洗滌三次和去離子水洗滌三次后,將洗滌后的反應產物用甲醇提取至三氯化鐵被完全洗掉,得到交聯多孔膜。交聯多孔膜吸收電解液后即得凝膠電解質。
實施例2
以PS25-PEO-PS25三段共聚物制備交聯多孔膜。
a)在無水無氧的環境下,將150ml的二氯甲烷和8.8g的聚氧化乙烯(Mn=8000g/mol)混合均勻后,向其中加入4.46ml的三乙胺并再混合均勻,得到第一混合液。
在無水無氧的環境下,將10mL的二氯甲烷和4.14mL的2-溴異丁酰溴混合均勻,得到第二混合液。
在冰水浴的溫度下,將第二混合液加入到第一混合液后得到反應體系,將反應體系在30℃的溫度下攪拌16h,得到第一中間體。使用飽和碳酸氫鈉溶液對第一中間體洗滌三次,再使用去離子水對第一中間體洗滌三次。向洗滌后的第一中間體內加入適量無水硫酸鈉進行干燥,過濾,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到冷的無水乙醚中沉降析出,過濾,得濾渣。用乙醚對濾渣洗滌多次后在32℃的溫度下烘干23h,得到Br-PEO-Br。
在無水無氧的環境下,將2g的Br-PEO-Br和0.072g的溴化亞銅混合均勻后,向其中加入20mL的1,4-二氧六環和1.45mL的苯乙烯單體混合均勻,得到反應體系。向反應體系內加入0.156mL的N,N,N',N,'N”-五甲基二亞乙基三胺并在105℃的溫度下反應25h,得到第二中間體。反應結束后將反應產物暴露在空氣中待冷卻至室溫后,向反應產物中加入四氫呋喃進行稀釋,得到稀釋液。將稀釋液過堿性三氧化二鋁短柱除去銅鹽,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到正己烷中沉降析出,過濾,得濾渣。將濾渣在40℃的溫度下烘干得到PS25-PEO-PS25三段共聚物。
b)將4g的聚偏氟乙烯(PVDF)和1g的PS25-PEO-PS25混合均勻后,向其中加入35.1mL的N,N-甲基吡咯烷酮,并在80℃的溫度下攪拌使形成均一的溶液后,向其中加入3.51mL的甘油,繼續在80℃的溫度下攪拌24h后,得到反應產物。將反應產物冷卻到室溫后脫泡。將脫泡后的反應產物澆鑄到培養皿中,在90℃的溫度下烘干4h后,再在溫度為120℃的真空環境中烘干24h,得到PVDF/PS25-PEO-PS25共混多孔膜。
c)在無水無氧的環境下,向4.875g的無水三氯化鐵中依次加入20mL的1,2-二氯乙烷和2.65mL的二甲氧基甲烷,得到反應體系。
在冰水浴的溫度下,將反映體系充分攪拌。向反應體系中加入500mg的PVDF/PS25-PEO-PS25共混多孔膜,在80℃的溫度下反應24h得到反應產物。將反應產物依次用丙酮洗滌3次、1mol/L的稀鹽酸溶液洗滌三次和去離子水洗滌三次后,將洗滌后的反應產物用甲醇提取至三氯化鐵被完全洗掉,得到交聯多孔膜。交聯多孔膜吸收電解液后即得凝膠電解質。
實施例3
以PS50-PEO-PS50三段共聚物制備交聯多孔膜。
a)在無水無氧的環境下,將150ml的二氯甲烷和8.8g的聚氧化乙烯(Mn=8000g/mol)混合均勻后,向其中加入4.46ml的三乙胺并再混合均勻,得到第一混合液。
在無水無氧的環境下,將10mL的二氯甲烷和4.14mL的2-溴異丁酰溴混合均勻,得到第二混合液。
在冰水浴的溫度下,將第二混合液加入到第一混合液后得到反應體系,將反應體系在25℃的溫度下攪拌20h,得到第一中間體。使用飽和碳酸氫鈉溶液對第一中間體洗滌三次,再使用去離子水對第一中間體洗滌三次。向洗滌后的第一中間體內加入適量無水硫酸鈉進行干燥,過濾,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到冷的無水乙醚中沉降析出,過濾,得濾渣。用乙醚對濾渣洗滌多次后在30℃的溫度下烘干24h,得到Br-PEO-Br。
在無水無氧的環境下,將2g的Br-PEO-Br和0.072g的溴化亞銅混合均勻后,向其中加入20mL的1,4-二氧六環和2.86mL的苯乙烯單體混合均勻,得到反應體系。向反應體系內加入0.156mL的N,N,N',N,'N”-五甲基二亞乙基三胺并在110℃的溫度下反應24h,得到第二中間體。將第二中間體暴露在空氣中待冷卻至室溫后,向反應產物中加入四氫呋喃進行稀釋,得到稀釋液。將稀釋液過堿性三氧化二鋁短柱除去銅鹽,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到正己烷中沉降析出,過濾,得濾渣。將濾渣在40℃的溫度下烘干得到PS50-PEO-PS50三段共聚物。
b)將4g的聚偏氟乙烯(PVDF)和1g的PS50-PEO-PS50混合均勻后,向其中加入35.1mL的N,N-甲基吡咯烷酮,并在80℃的溫度下攪拌使形成均一的溶液后,向其中加入3.51mL的甘油,繼續在80℃的溫度下攪拌24h后,得到反應產物。將反應產物冷卻到室溫后脫泡。將脫泡后的反應產物澆鑄到培養皿中,在90℃的溫度下烘干4h后,再在溫度為120℃的真空環境中烘干24h,得到PVDF/PS50-PEO-PS50共混多孔膜。
c)在無水無氧的環境下,向4.875g的無水三氯化鐵中依次加入20mL的1,2-二氯乙烷和2.65mL的二甲氧基甲烷,得到反應體系。
在冰水浴的溫度下,將反映體系充分攪拌。向反應體系中加入500mg的PVDF/PS50-PEO-PS50共混多孔膜,在80℃的溫度下反應24h得到反應產物。將反應產物依次用丙酮洗滌3次、1mol/L的稀鹽酸溶液洗滌三次和去離子水洗滌三次后,將洗滌后的反應產物用甲醇提取至三氯化鐵被完全洗掉,得到交聯多孔膜。交聯多孔膜吸收電解液后即得凝膠電解質。
實施例4
以PS100-PEO-PS100三段共聚物制備交聯多孔膜。
a)在無水無氧的環境下,將150ml的二氯甲烷和8.8g的聚氧化乙烯(Mn=8000g/mol)混合均勻后,向其中加入4.46ml的三乙胺并再混合均勻,得到第一混合液。
在無水無氧的環境下,將10mL的二氯甲烷和4.14mL的2-溴異丁酰溴混合均勻,得到第二混合液。
在冰水浴的溫度下,將第二混合液加入到第一混合液后得到反應體系,將反應體系在30℃的溫度下攪拌16h,得到第一中間體。使用飽和碳酸氫鈉溶液對第一中間體洗滌三次,再使用去離子水對第一中間體洗滌三次。向洗滌后的第一中間體內加入適量無水硫酸鈉進行干燥,過濾,得濾液。將濾液中的DMC蒸發至少量后,將濾液加入到冷的無水乙醚中沉降析出,過濾,得濾渣。用乙醚對濾渣洗滌多次后在30℃的溫度下烘干24h,得到Br-PEO-Br。
在無水無氧的環境下,將2g的Br-PEO-Br和0.072g的溴化亞銅混合均勻后,向其中加入20mL的1,4-二氧六環和5.71mL的苯乙烯單體混合均勻,得到反應體系。向反應體系內加入0.156mL的N,N,N',N,'N”-五甲基二亞乙基三胺并在110℃的溫度下反應24h,得到第二中間體。將第二中間體暴露在空氣中待冷卻至室溫后,向反應產物中加入四氫呋喃進行稀釋,得到稀釋液。將稀釋液過堿性三氧化二鋁短柱除去銅鹽,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到正己烷中沉降析出,過濾,得濾渣。將濾渣在40℃的溫度下烘干得到PS100-PEO-PS100三段共聚物。
b)將4g的聚偏氟乙烯(PVDF)和1g的PS100-PEO-PS100混合均勻后,向其中加入35.1mL的N,N-甲基吡咯烷酮,并在80℃的溫度下攪拌使形成均一的溶液后,向其中加入3.51mL的甘油,繼續在80℃的溫度下攪拌24h后,得到反應產物。將反應產物冷卻到室溫后脫泡。將脫泡后的反應產物澆鑄到培養皿中,在90℃的溫度下烘干5h后,再在溫度為125℃的真空環境中烘干23h,得到PVDF/PS100-PEO-PS100共混多孔膜。
c)在無水無氧的環境下,向4.875g的無水三氯化鐵中依次加入20mL的1,2-二氯乙烷和2.65mL的二甲氧基甲烷,得到反應體系。
在冰水浴的溫度下,將反映體系充分攪拌。向反應體系中加入500mg的PVDF/PS100-PEO-PS100共混多孔膜,在80℃的溫度下反應24h得到反應產物。將反應產物依次用丙酮洗滌三次、1mol/L的稀鹽酸溶液洗滌三次和去離子水洗滌三次后,將洗滌后的反應產物用甲醇提取至三氯化鐵被完全洗掉,得到交聯多孔膜。交聯多孔膜吸收電解液后即得凝膠電解質。
實施例5
以PS230-PEO-PS230三段共聚物制備交聯多孔膜。
按照實施例4的方案制得Br-PEO-Br。
在無水無氧的環境下,將2g的Br-PEO-Br和0.072g的溴化亞銅混合均勻后,向其中加入20mL的1,4-二氧六環和11.43mL的苯乙烯單體混合均勻,得到反應體系。向反應體系內加入0.156mL的N,N,N',N,'N”-五甲基二亞乙基三胺并在110℃的溫度下反應22h,得到第二中間體。將第二中間體暴露在空氣中待冷卻至室溫后,向反應產物中加入四氫呋喃進行稀釋,得到稀釋液。將稀釋液過堿性三氧化二鋁短柱除去銅鹽,得濾液。將濾液中的溶劑蒸發至少量后,將濾液加入到正己烷中沉降析出,過濾,得濾渣。將濾渣在40℃的溫度下烘干得到PS230-PEO-PS230三段共聚物。
b)將4g的聚偏氟乙烯(PVFD)和1g的PS230-PEO-PS230混合均勻后,向其中加入35.1mL的N,N-甲基吡咯烷酮,并在80℃的溫度下攪拌使形成均一的溶液后,向其中加入3.51mL的甘油,繼續在80℃的溫度下攪拌24h后,得到反應產物。將反應產物冷卻到室溫后脫泡。將脫泡后的反應產物澆鑄到培養皿中,在90℃的溫度下烘干4.5h后,再在溫度為110℃的真空環境中烘干24h,得到PVDF/PS230-PEO-PS230共混多孔膜。
c)在無水無氧的環境下,向4.875g的無水三氯化鐵中依次加入20mL的1,2-二氯乙烷和2.65mL的二甲氧基甲烷,得到反應體系。
在冰水浴的溫度下,將反映體系充分攪拌。向反應體系中加入500mg的PVDF/PS230-PEO-PS230共混多孔膜,在80℃的溫度下反應22h得到反應產物。將反應產物依次用丙酮洗滌三次、1mol/L的稀鹽酸溶液洗滌三次和去離子水洗滌三次后,將洗滌后的反應產物用甲醇提取至三氯化鐵被完全洗掉,得到交聯多孔膜。交聯多孔膜吸收電解液后即得凝膠電解質。
承上述,將實施例1制備的PS15-PEO-PS15即為CPE-1,實施例2制備的PS25-PEO-PS25即為CPE-2,實施例3制備的PS50-PEO-PS50即為CPE-3,實施例4制備的PS100-PEO-PS100即為CPE-4,實施例5制備的PS230-PEO-PS230即為CPE-5。測量或統計CPE-1、CPE-2、CPE-3、CPE-4和CPE-5的分子量、分子量分布和組成,其結果見表1。
表1 PS-PEO-PS三段共聚物的分子量、分子量分布以及組成
從表1中可以看出,本PDI(聚合物分散性指數)為分子量分布。CPE-1、CPE-2、CPE-3、CPE-4和CPE-5的PDI數值均較小,其分子量分布較窄。
對比例
以聚偏氟乙烯(PVDF)以相轉化法制備多孔膜。將5g的聚偏氟乙烯和35.1mL的N,N-甲基吡咯烷酮,并在80℃的溫度下攪拌使形成均一的溶液后,向其中加入3.51mL的甘油,繼續在80℃的溫度下攪拌24h后,得到反應產物。將反應產物冷卻到室溫后脫泡。將脫泡后的反應產物澆鑄到培養皿中,在90℃的溫度下烘干5h后,再在溫度為125℃的真空環境中烘干23h,得到PVDF共混多孔膜。對比例即為PVDF。可以理解,
分別測量PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的孔隙率(Porosity)、吸液率(Electrolyte uptake)、離子電導率(Ionic conductivity)和鋰離子傳導活化能(Activation energy),以及分別進行BET測試,其結果見表2。
表2性能測試結果
從表2可以看出,通過交聯制備的CPE-1、CPE-2、CPE-3、CPE-4和CPE-5的孔隙度率、吸液率、離子電導率、鋰離子傳導活化能和BET性能均相比單一的PVDF多孔膜有顯著提高。本發明實施例1~5制備的交聯多孔膜具有較好的孔隙分布、較強的吸液能力和優良的導電性能。尤其通過BET測試可以看出,隨著交聯多孔膜中嵌段共聚物中PS鏈段長度的增加,其交聯度提高明顯。
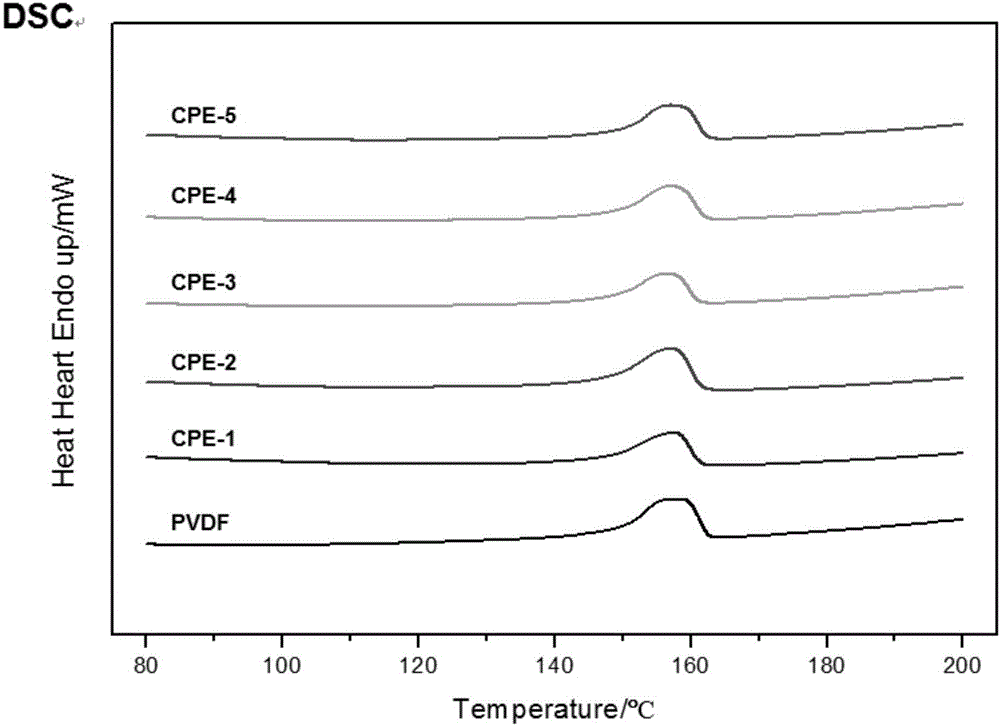
分別對PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別進行膜的熱分析,包括DSC分析和TGA分析,其結果見圖1和圖2。
從圖1中可以看出,DSC在160℃及其附近溫度存在PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的熔融峰。可以理解,對照組和實施例均包含相同的組成聚偏氟乙烯,該熔融峰為聚偏氟乙烯的熔融峰。
從圖2中可以看出,CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別在300℃及其附近溫度開始熱分解。
分別對CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別進行膜經過不同溫度處理后的孔隙率分析,其結果見圖3。
從圖3中可以看出,CPE-1 precursor指的是交聯多孔膜前體,即為相轉化法制得的交聯前的共混多孔膜,CPE-2 precursor、CPE-3 precursor、CPE-4 precursor和CPE-5 precursor同理。可以理解,沒有交聯的共混多孔膜在180℃時發生熔融閉孔無法使用。交聯多孔膜在160℃時孔隙率幾乎沒有下降;溫度繼續升高到200℃時,其孔隙率最高保持在40%左右;溫度升高到260℃時,其孔隙率最高保持在30%左右。
在不同的溫度下,分別對PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別進行膜的熱收縮性測試,其結果見圖4。
從圖4可以看出,制備的CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的交聯多孔膜在180℃和260℃的高溫下熱處理1h尺寸收縮非常小,而對照組的PVDF在180℃時即會發生迅速熔融。同時,對比商品化的PP隔膜,同等條件下可以看出,商品化的PP隔膜在180℃時發生了嚴重尺寸收縮,在260℃時完全降解。因此,本發明實施例制備的交聯多孔膜在高溫下具有優異的尺寸穩定性,從而提高了鋰離子電池的安全性能。
承上述,本交聯多孔膜具備較好的熱穩定性,能夠保證鋰離子電池在較苛刻的條件下正常使用和安全使用。
在20~25℃的溫度下,分別對PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的交聯多孔膜進行交流阻抗測試,其結果見圖5。
從圖5中可以看出,PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的凝膠電解質具有在常溫下具有較低的交流阻抗。
分別對PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的交聯多孔膜進行線性伏安掃描曲線測試,電化學窗口為5V,其結果見圖6。
分別對PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的交聯多孔膜組裝成Li/LiFePO4半電池進行充放電測試。以電流密度為0.1C,在2.5~4.2V循環40次,其結果見圖7。
從圖7中可以看出,PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的初始容量分別為125.3mA·h/g、145.3mA·h/g、138.4mA·h/g、133.8mA·h/g、140.4mA·h/g和128.2mA·h/g。循環40次后,PVDF、CPE-1、CPE-2、CPE-3、CPE-4和CPE-5組別的容量保留率分別為78.1%、95.5%、100%、97.5%、90.7%和87.7%。本發明實施例制備電解質的容量保留率相比于對照組均有顯著提高,具有更長使用壽命。
承上述,本發明實施例制備的凝膠電解質在140℃高溫下保持很高的離子電導率(7.61×10-3S/cm),而對照組裝成電池測試時在140℃時就已經閉孔,進而無法測試。
以上所描述的實施例是本發明一部分實施例,而不是全部的實施例。本發明的實施例的詳細描述并非旨在限制要求保護的本發明的范圍,而是僅僅表示本發明的選定實施例。基于本發明中的實施例,本領域普通技術人員在沒有作出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 摻雜碳的凝膠電解液、生產方法及在芯包上的形成方法與流程
- 非水系電解液的制造方法與工藝
- 非水系電解質二次電池用正極活性物質、其制造方法和使用其的非水系電解質二次電池與流程
- 非水系電解質二次電池用的正極活性物質及其制造方法、以及非水系電解質二次電池與流程
- 非水系電解質二次電池用正極活性物質和其制造方法、以及使用了該正極活性物質的非水系電解質二次電池與流程
- 非水系電解質二次電池用正極活性物質和其制造方法,以及使用了該正極活性物質的非水系電解質二次電池與流程
- 非水系電解液及使用該電解液的非水系電解液二次電池的制造方法與工藝
- 非水系電解質二次電池用正極活性物質和其制造方法,以及使用了該正極活性物質的非水系電解質二次電池與流程
- 非水系電解質二次電池用正極活性物質和其制造方法、及非水系電解質二次電池與流程
- 非水系電解液和使用了該非水系電解液的非水系電解液二次電池的制造方法與工藝