一種磷酸鐵鋰正極材料的制備方法與流程
本發明涉及電池材料技術領域,尤其涉及一種磷酸鐵鋰正極材料的制備方法。
背景技術:
磷酸鐵鋰能夠可逆地嵌入和脫嵌鋰離子,具有原材料來源豐富、比容量高、循環壽命長、安全無毒、環境友好等特點,被認為是新能源動力電池的理想正極材料。
然而,磷酸鐵鋰電導率低,導電性差,極片在充放電過程中容易產生極化,低溫環境中,Li+在活性物質中遷移路徑較長,遷移速率慢,遷移過程中所受阻力較大,低溫充放電性能差;此外,電流密度增大時,Li+在活性物質遷移所受阻力增大,極化加劇,電池倍率性能急劇下降,使其在低溫環境中應用受到限制。
目前,對磷酸鐵鋰改性的方法主要有三種:一、利用金屬離子對磷酸鐵鋰進行摻雜,提高材料本征電導率,降低極化,但此方法但并不能解決Li+遷移速率慢的問題,因此極片極化仍然嚴重;二、利用碳材料對磷酸鐵鋰進行包覆,增大磷酸鐵鋰顆粒之間電導率,并改善顆粒表面各相異性,降低極片極化,并保證Li+順利遷移,然而,目前所使用碳材料多為無定形碳,孔結構無序,孔徑分布不均,無法解決大電流密度下低溫電池倍率性能差的問題;三、將磷酸鐵鋰納米化,縮短Li+遷移路徑,但無法解決磷酸鐵鋰電導率低等根本性問題。
鑒于此,實有必要提供一種新型的磷酸鐵鋰正極材料的制備方法以克服以上缺陷。
技術實現要素:
本發明的目的是提供一種具有較好低溫充放電性能及低溫倍率性能的磷酸鐵鋰正極材料的制備方法。
為了實現上述目的,本發明提供一種磷酸鐵鋰正極材料的制備方法,包括如下步驟:步驟一:按照一定質量比稱取石墨、濃硝酸和濃磷酸混合,置于水浴中攪拌,溫度升高至80-90℃,按照一定質量比加入雙氧水,攪拌,將產物降溫至室溫,水洗并抽濾,將抽濾后的濾餅進行干燥,得到氧化石墨GO;步驟二:按照一定質量比稱取碳酸鋰、碳酸鈉和碳酸鉀,混合并進行球磨,對球磨后的混合物進行加熱,插入鐵絲和Ni-Cr合金絲,通入CO2,通電進行電解,利用鹽酸和去離子水對電解產物進行交替清洗,直至清洗后液體呈中性,烘干,得到蜂窩狀碳材料HC;步驟三:按照一定質量比稱取磷酸鐵鋰和氧化石墨GO溶于無水乙醇中,將混合物置于微波反應器中保持一段時間后,按照一定質量比加入蜂窩狀碳材料HC,轉移至高壓反應釜中,進行水熱反應,將水熱反應后的產物進行水洗、抽濾及干燥,得到改性的磷酸鐵鋰材料LiFePO4/GO/HC。
在一個優選實施方式中,所述步驟一中的石墨、濃硝酸和濃磷酸的質量比為1:6:4-1:6:6。
在一個優選實施方式中,所述步驟一中的雙氧水與石墨質量比為1:2-1:2.5。
在一個優選實施方式中,所述步驟一中濾餅的干燥溫度為90-100℃,干燥時間為6-8h。
在一個優選實施方式中,所述步驟二中的碳酸鋰、碳酸鈉和碳酸鉀的質量比為4:1:1-5:1:1。
在一個優選實施方式中,所述步驟二中進行電解的電流密度為200-300mA/cm3。
在一個優選實施方式中,所述步驟二中的鹽酸的濃度為1%-2%。
在一個優選實施方式中,所述步驟三中的磷酸鐵鋰與氧化石墨GO的質量比為10:1-12:1。
在一個優選實施方式中,所述步驟三中的磷酸鐵鋰與蜂窩狀碳材料HC的質量比為10:0.2-10:0.3。
在一個優選實施方式中,所述步驟三中的水熱反應溫度為110-120℃,水熱反應時間為20-24h。
相比于現有技術,本發明提供的磷酸鐵鋰正極材料的制備方法,所制備的正極材料具有較好低溫充放電性能及低溫倍率性能。
【附圖說明】
圖1為本發明實施例1制備的氧化石墨GO、及改性的磷酸鐵鋰材料LiFePO4/GO/HC的XRD圖;
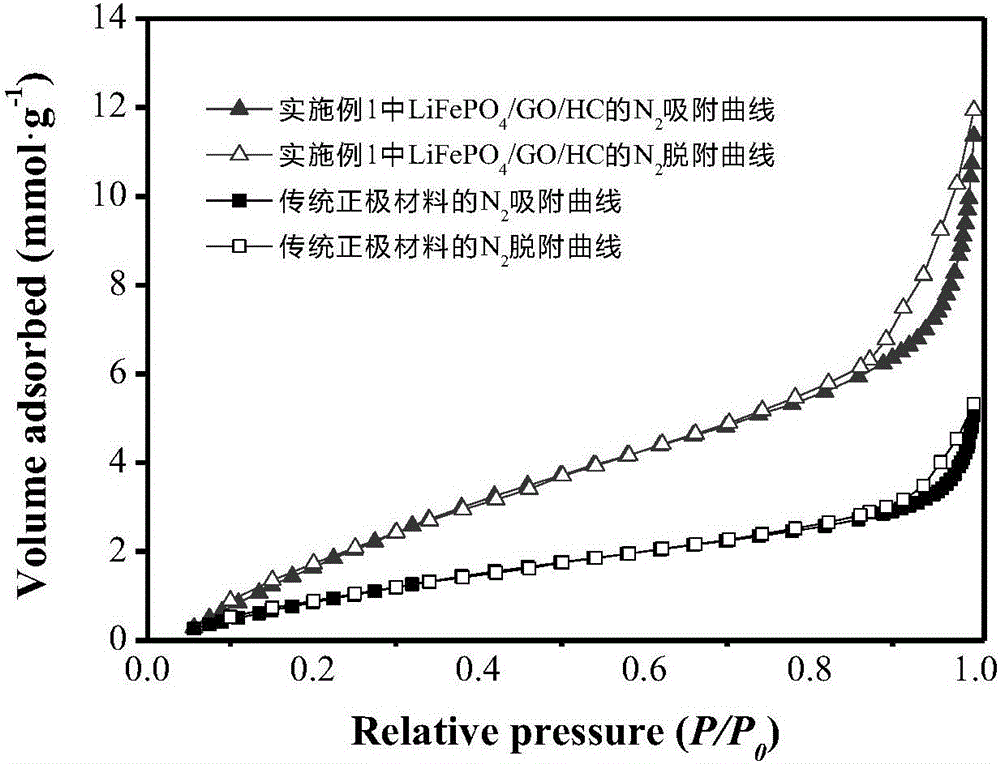
圖2為傳統碳材料及本發明實施例1所制得蜂窩狀碳材料HC的N2吸附-脫附曲線圖;
圖3為本發明實施例1所制得的蜂窩狀碳材料HC的SEM圖。
【具體實施方式】
為了使本發明的目的、技術方案和有益技術效果更加清晰明白,以下結合附圖和具體實施方式,對本發明進行進一步詳細說明。應當理解的是,本說明書中描述的具體實施方式僅僅是為了解釋本發明,并不是為了限定本發明。
本發明提供一種磷酸鐵鋰正極材料的制備方法,包括如下步驟:
步驟一:按照一定質量比稱取石墨、濃硝酸和濃磷酸混合,置于水浴中攪拌,溫度升高至80-90℃,按照一定質量比加入雙氧水,攪拌,將產物降溫至室溫,水洗并抽濾,將抽濾后的濾餅進行干燥,得到氧化石墨GO;
步驟二:按照一定質量比稱取碳酸鋰、碳酸鈉和碳酸鉀,混合并進行球磨,對球磨后的混合物進行加熱,插入鐵絲和Ni-Cr合金絲,通入CO2,通電進行電解,利用鹽酸和去離子水對電解產物進行交替清洗,直至清洗后液體呈中性,烘干,得到蜂窩狀碳材料HC;
步驟三:按照一定質量比稱取磷酸鐵鋰和氧化石墨GO溶于無水乙醇中,將混合物置于微波反應器中保持一段時間后,按照一定質量比加入蜂窩狀碳材料HC,轉移至高壓反應釜中,進行水熱反應,將水熱反應后的產物進行水洗、抽濾及干燥,得到改性的磷酸鐵鋰材料LiFePO4/GO/HC,即最終的磷酸鐵鋰正極材料。
具體的,所述步驟一中的石墨、濃硝酸和濃磷酸的質量比為1:6:4-1:6:6,其中,所述濃硝酸和濃磷酸均為溶液,所述濃硝酸摩爾濃度為14.5mol/L,即質量分數為65%,所述濃磷酸摩爾濃度為14.75mol/L,即質量分數85.5%。所述步驟一中的雙氧水與石墨質量比為1:2-1:2.5。所述步驟一中濾餅的干燥溫度為90-100℃,干燥時間為6-8h。
具體的,所述步驟二中的碳酸鋰、碳酸鈉和碳酸鉀的質量比為4:1:1-5:1:1,其中,對碳酸鋰、碳酸鈉和碳酸鉀進行加熱后,三中碳酸鹽在高溫下熔化形成離子熔體即熔融鹽,熔融態的碳酸鹽具有較好的導電性,能夠作為電解質。所述步驟二中進行電解的電流密度為200-300mA/cm3,電解時,鐵絲和Ni-Cr合金分別作為電解池中陰極和陽極,進行導電,在高溫下,電解過程中發生氧化還原過程:CO2在陰極表面還原得到蜂窩狀碳材料HC,在陽極表面氧化得到氧氣。所述步驟二中的鹽酸的濃度為1%-2%。
具體的,所述步驟三中的磷酸鐵鋰與氧化石墨GO的質量比為10:1-12:1。所述步驟三中的磷酸鐵鋰與蜂窩狀碳材料HC的質量比為10:0.2-10:0.3。所述步驟三中的水熱反應溫度為110-120℃,水熱反應時間為20-24h。其中,磷酸鐵鋰為尖晶石結構,其中磷酸亞鐵離子與磷酸根離子以頭對尾方式進行接觸,導電性差,鋰離子分散于晶體空隙之中,氧化石墨GO為層狀物質,利用氧化石墨GO與磷酸鐵鋰混合,經過步驟3,氧化石墨片插入磷酸亞鐵離子與磷酸根離子之間,提高了最終的正極材料的導電性及正極材料與電解液兼容性,提高了充放電性能。其中,微波反應器傳熱好,受熱均勻,蜂窩狀碳材料HC包覆效果好,且微波反應器反應速度快,可以縮短制備周期。
實施例1:
1、稱取10g石墨、60g濃硝酸和40g濃磷酸混合,于0℃水浴中攪拌6h,將溫度升高至90℃,加入20g雙氧水,攪拌2h,將產物降溫至室溫,水洗并抽濾,將濾餅置于100℃中空干燥箱中干燥6h,得到氧化石墨GO;
2、稱取8g碳酸鋰、2g碳酸鈉和2g碳酸鉀混合并進行球磨至200nm,將球磨后的混合物置于陶瓷加熱器中,蓋上電解槽蓋,并加熱至550℃后,插入鐵絲和Ni-Cr合金絲,通入CO2,通電進行電解,電解的電流密度為200mA/cm3,使碳納米管的粗產物在鐵絲表面沉積;利用1%的鹽酸和去離子水對碳納米管的粗產物進行交替清洗,直至清洗后液體呈中性,將所得產物置于100℃干燥箱中干燥4h,得到蜂窩狀碳材料HC;
3、稱取25g磷酸鐵鋰和2.5g氧化石墨GO溶于320ml無水乙醇中,將反應物置于微波反應器中保持3h后,加入0.5g步驟2制得的蜂窩狀碳材料HC,將混合物轉移至高壓反應釜中,置于110℃干燥箱中進行水熱處理24h,將所得產物進行水洗、抽濾,將所得產物置于110℃干燥箱中干燥8h,得到改性的磷酸鐵鋰材料LiFePO4/GO/HC。
實施例2:
1、稱取10g石墨、60g濃硫酸和40g濃磷酸混合,于0℃水浴中攪拌6h,將溫度升高至90℃,加入20g雙氧水,攪拌2h,將產物降溫至室溫,水洗并抽濾,將濾餅置于100℃中空干燥箱中干燥6h,得到氧化石墨GO;
2、稱取8g碳酸鋰、2g碳酸鈉和2g碳酸鉀混合并進行球磨至200nm,將球磨后的混合物置于陶瓷加熱器中,蓋上電解槽蓋,并加熱至600℃后,插入鐵絲和Ni-Cr合金絲,通入CO2,通電進行電解,電解的電流密度為200mA/cm3,使碳納米管的粗產物在鐵絲表面沉積;利用1%的鹽酸和去離子水對碳納米管的粗產物進行交替清洗,直至清洗后液體呈中性,將所得產物置于100℃干燥箱中干燥4h,得到蜂窩狀碳材料HC;
3、稱取25g磷酸鐵鋰和2.5g氧化石墨GO溶于320ml無水乙醇中,將反應物置于微波反應器中保持3h后,加入0.5g驟2制得的蜂窩狀碳材料HC,將混合物轉移至高壓反應釜中,置于110℃干燥箱中進行水熱處理24h,將所得產物進行水洗、抽濾,將所得產物置于110℃干燥箱中干燥8h,得到改性的磷酸鐵鋰材料LiFePO4/GO/HC。
實施例3:
1、稱取10g石墨、60g濃硝酸和40g濃硫酸混合,于0℃水浴中攪拌6h,將溫度升高至90℃,加入20g雙氧水,攪拌2h,將產物降溫至室溫,水洗并抽濾,將濾餅置于100℃中空干燥箱中干燥6h,得到氧化石墨GO;
2、稱取8g碳酸鋰、2g碳酸鈉和2g碳酸鉀混合并進行球磨至200nm,將球磨后的混合物置于陶瓷加熱器中,蓋上電解槽蓋,并加熱至600℃后,插入鐵絲和Ni-Cr合金絲,通入CO2,通電進行電解,電解的電流密度為300mA/cm3,使碳納米管的粗產物在鐵絲表面沉積;利用1%的鹽酸和去離子水對碳納米管的粗產物進行交替清洗,直至清洗后液體呈中性,將所得產物置于100℃干燥箱中干燥4h,得到蜂窩狀碳材料HC;
3、稱取25g磷酸鐵鋰和2.5g氧化石墨GO溶于320ml無水乙醇中,將反應物置于微波反應器中保持3h后,加入0.5g步驟2制得的蜂窩狀碳材料HC,將混合物轉移至高壓反應釜中,置于110℃干燥箱中進行水熱處理24h,將所得產物進行水洗、抽濾,將所得產物置于110℃干燥箱中干燥8h,得到改性磷酸鐵鋰材料LiFePO4/GO/HC。
正極極片的制備及扣式電池的組裝與測試:
正極分別以傳統方法制備的碳包覆的磷酸鐵鋰和本發明實施例1制備的改性的磷酸鐵鋰材料LiFePO4/GO/HC為活性物質,Super-P為導電極,聚偏氟乙烯(PVDF)為粘結劑,依次按照94:3:3的比例與N-甲基吡咯烷酮(NMP)混合并攪拌均勻后得到漿料。負極材料選用人造石墨(D50為8.39μm),電解液選用電解質為LiPF6的低溫電解液,隔膜選用16μm厚PP隔膜。經過配料、涂布、棍壓、制片、卷繞、裝配、注液、封口等工序制成圓柱鋼殼型電池。
傳統方法制備的碳包覆的磷酸鐵鋰正極的電池編號為A,本發明實施例1制備的改性的磷酸鐵鋰材料LiFePO4/GO/HC正極的電池編號為B。
測定電池A及電池B的低溫充放電性能及循環性能,充放電截止電壓范圍2.0~3.6V,測試溫度-20℃。
表1為傳統方法制備的碳包覆的磷酸鐵鋰正極的電池A與本發明實施例1制備的改性的磷酸鐵鋰材料LiFePO4/GO/HCB正極的電池B在-20℃下的充放電性能對照表:
表1:
由表1可知,電流密度分別由0.5C增大至1C時,A、B兩支電池充電恒流比和放電比率均逐漸減小。增大電流密度,極片極化增大,充放電性能降低,因此充電恒流比和放電比率均減小。然而,實驗條件下,電池A的充電恒流比和放電比率電壓均明顯低于電池B,這是由于本發明實施例1中改性的磷酸鐵鋰材料LiFePO4/GO/HC含有氧化石墨,可以提高正極活性物質電導率,并增大Li+遷移速率,降低極化。同時,活性物質外表面包覆的蜂窩狀碳材料可以提高活性物質與電解液浸潤性,并且可以接收從各個方向擴散的Li+,有利于Li+嵌入。此外,包覆層碳材料孔徑較大,可以保證大電流密度下Li+順利嵌入與脫出,提高電池低溫倍率性能。
圖1為本發明實施例1制備的氧化石墨GO、及改性的磷酸鐵鋰材料LiFePO4/GO/HC的XRD圖,由圖1可以看出,改性后的磷酸鐵鋰材料中出現氧化石墨GO的對應峰,說明氧化石墨GO被成功引入至磷酸鐵鋰中,利用氧化石墨GO對磷酸鐵鋰進行改姓,有利于提高磷酸鐵鋰導電性,降低極化,并且有利于提高Li+遷移速率。
圖2為傳統碳材料及本發明實施例1所制得蜂窩狀碳材料HC的N2吸附-脫附曲線圖,由圖2可以看出,吸附-脫附曲線呈第Ⅲ型吸附等溫線,并且含有回滯環,傳統碳材料及本發明實施例1所制得蜂窩狀碳材料HC的比表面積分別為1124m2/g和1942m2/g,平均孔徑分別為7nm和13nm;與傳統碳材料相較,本發明實施例1中的蜂窩狀碳材料HC具有更大的比表面積和孔徑,可以使Li+更容易實現嵌入/脫出,更容易通過碳材料進入至活性中心,提高電池低溫性能。
圖3為本發明實施例1所制得的蜂窩狀碳材料HC的SEM圖,由圖3可以看出,本發明中實施例1中的蜂窩狀碳材料HC呈蜂窩狀結構,利用此蜂窩狀碳材料HC對磷酸鐵鋰進行改性,有利于增大電解液浸潤性,并且有利于Li+更容易從各個方向進行嵌入/脫出,提高電池充放電性能。
本發明提供的磷酸鐵鋰正極材料的制備方法,以石墨為碳源,利用濃硝酸、濃磷酸和雙氧水對石墨進行氧化,制備氧化石墨GO;以CO2為碳源,通過調節溫度及電流密度制備具有較大比表面積和孔徑并且呈蜂窩狀的碳材料HC,通過調節電流密度可以調控蜂窩狀碳材料的孔徑;以所制得的氧化石墨GO和蜂窩狀碳材料HC對磷酸鐵鋰進行改性,制得石墨插層、碳材料包覆的正極材料LiFePO4/GO/HC,提高了材料導電性,使Li+在遷移更短的距離后即可到達導電性較好的氧化石墨GO表面,增大遷移速率;碳材料呈蜂窩狀,可以增大電解液浸潤性,并且有利于Li+更容易從各個方向進行嵌入/脫出,提高電池充放電性能;并且,蜂窩狀碳材料HC的比表面積較大,孔徑較大,可以保證更多Li+進入碳材料后容易通過碳材料孔道進入至活性中心,提高材料低溫倍率性能,同時減小Li+在材料表面沉積,提高電池安全性。
本發明提供的磷酸鐵鋰正極材料的制備方法所制備的正極材料具有較好低溫充放電性能及低溫倍率性能。
本發明并不僅僅限于說明書和實施方式中所描述,因此對于熟悉領域的人員而言可容易地實現另外的優點和修改,故在不背離權利要求及等同范圍所限定的一般概念的精神和范圍的情況下,本發明并不限于特定的細節、代表性的設備和這里示出與描述的圖示示例。
- 還沒有人留言評論。精彩留言會獲得點贊!