一種超電容用金屬離子摻雜花狀MnO2納米片及其制備方法與流程
本發明涉及超電容電極材料的制備領域,具體涉及一種超電容用金屬離子摻雜花狀MnO2納米片及其制備方法。
背景技術:
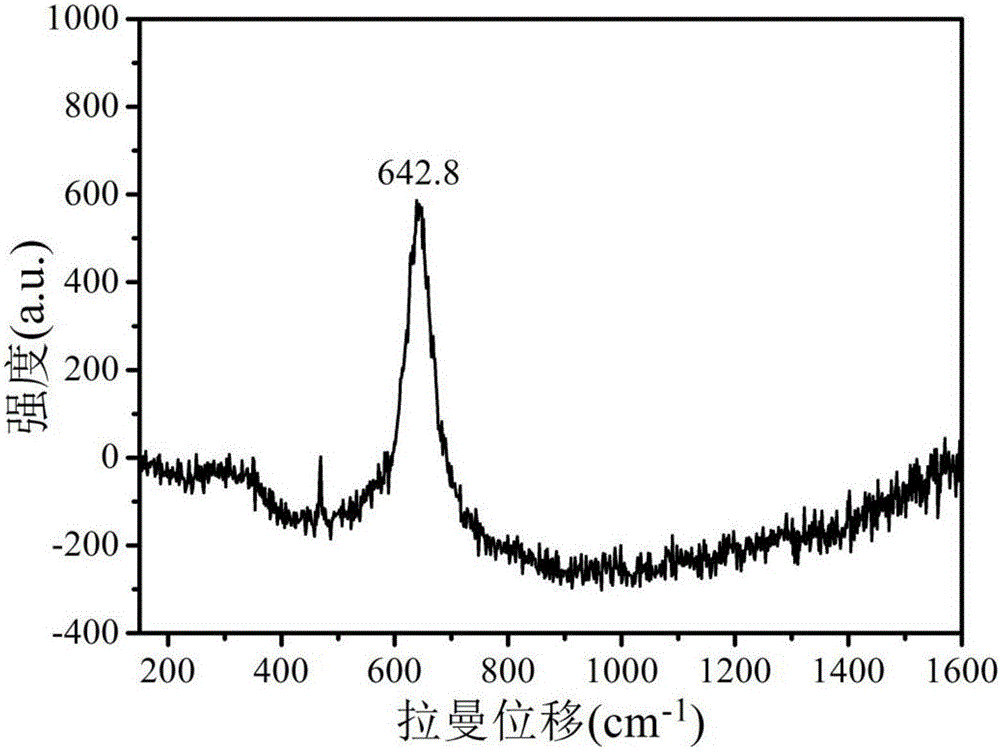
:二氧化錳作為過渡金屬氧化物,存在可變的氧化價態,具有價格低廉、環境友好、來源廣泛等優點,同時其優異的理論比電容特性,使其作為超級電容器電極材料具有很大的發展潛力。但是,二氧化錳作為電極材料時較差的導電性和電極穩定性,導致其實際比電容值遠低于其理論值。提高導電性及電極穩定性已成為亟待解決的問題。隨著納米材料及摻雜技術的發展,具有特殊納米結構或者摻雜金屬氧化物因具有高比表面積、多種氧化價態和高比電容量等特點已成為國內外研究熱點。FengXM等[JournalofMaterialsChemistryA,2013,1,12818-12825]通過一步水熱法實現了形貌可控的二氧化錳/石墨烯復合材料的制備,制得的花瓣狀二氧化錳納米片/石墨烯復合材料的比電容值高達516.8F/g,且具有良好的循環穩定性。DaiYuming等[ChemistryLetters,2014,43(1):122-124.]在磁力攪拌的情況下,將水熱法制備的MnO2納米線加入到HAuCl4溶液中(40mL,1mmol/L),通過加入0.06mmol抗壞血酸,制得Au包裹MnO2納米線的復合電極材料,其比電容值可達400F/g。但這兩種方法制備流程復雜,制備過程難以有效控制,且部分藥品價格較貴,難以進行工業推廣。公開號為CN104310486A的專利文獻公開了一種一步合成單層二氧化錳納米片的方法,該方法通過高錳酸鹽在酸性烷基硫酸鹽表面活性劑水溶液中的一步反應,利用烷基硫酸鹽表面活性劑水解生成相應的醇原位還原高錳酸鹽生成二氧化錳,但該方法制備過程簡單,能夠得到超薄納米片結構且具有良好的氧化還原特性,但是該方法使用的烷基表面活性劑具有毒性,且使用的酸溶液均為強酸,對環境破壞較為嚴重,該方法不利于商業推廣。公開號為CN103641174A的專利文獻公開了一種納米片狀MnO2-石墨烯復合材料的制備方法,該方法為:硝酸錳溶液在雙氧水和堿混合溶液中進行氧化還原反應制得粉體;將所述粉體分散于過硫酸銨溶液處理,然后分散到有機溶劑中,得到納米片狀MnO2溶液;將所述納米片狀MnO2溶液稀釋后,與石墨烯粉體的分散液混合,超聲處理制得到納米片狀MnO2-石墨烯復合材料,其比電容值可達578F/g,但該方法制備流程復雜,且MnO2與石墨烯為物理結合不利于電極穩定性的提升。技術實現要素:本發明的目的是提供一種超電容用金屬離子摻雜花狀MnO2納米片及其制備方法,該方法制備流程短、成本低、無污染且電化學特性優異。為達到上述目的,本發明提供的技術方案是:一種超電容用金屬離子摻雜花狀MnO2納米片的制備方法,包括如下步驟:S1、配制濃度比為1:1的硫酸錳和過硫酸銨混合水溶液,將其置于超聲恒溫水浴環境中,同時將金屬鹽溶液勻速滴加到混合溶液中,隨后繼續反應一段時間;S2、將反應產物離心洗滌后放入蒸發皿中,干燥后,放入保濕箱中留存。步驟S1中,制備溫度為30~60℃,反應時間為0.5~3h。步驟S1中,所滴加的金屬鹽為鎳、鋁、鋅或錫的硫酸鹽溶液中的一種,滴加時間為2~5min。步驟S1中,所滴加金屬鹽溶液濃度為0.005~0.1mol/L。步驟S2中,干燥條件為30~50℃干燥2~5h,或者-30℃~-50℃冷凍干燥5~10h。本發明制得的MnO2納米片自組裝成花狀。優選的,步驟S1中硫酸錳和過硫酸銨的濃度為1mol/L,滴加的硫酸鎳溶液濃度為0.005mol/L;優選的,步驟S1中所滴加的金屬鹽溶液為鎳、鋁、鋅及錫金屬硫酸鹽的一種,滴加時間為3min,反應體系溫度為40℃,間為1h;優選的,步驟S2的干燥方式為-30℃冷凍干燥5h。在制備起始階段,MnO2納米片形核長大,隨著摻雜離子的加入,離子的聚集效應使得MnO2納米片局部區域產生成分起伏和能量起伏從而導致新的形核點形成,在此基礎上生長為納米片,通過這種生長方式,使得MnO2呈現花瓣狀的形貌。這種發達花片狀形貌有效抑制了MnO2納米片的重疊和聚集,使其具有穩定的三維結構和較大的比表面積。其作為電極材料工作時能夠具有較大的活性面積,同時電解液與電極材料充分接觸有利于離子在電極體系中快速高效地交換傳輸。因此,該方法制得的超級電容器電極材料具有優異的電化學特性。本發明公開了超電容用金屬離子摻雜花狀MnO2納米片及其制備方法,采用化學沉淀的方法通過摻雜金屬離子引導制備納米片自組裝花狀的MnO2,其片層厚度為3~5nm。本發明制備流程短、效率高且成本低、環境污染小,制得的花狀MnO2電極材料具有優異的電化學特性;該反應體系簡單,且容易控制反應進程,制備的花狀MnO2具有三維立體結構因而其電極活性面積較大,有利于電解質離子在電極體相中快速傳輸,明顯提升電化學性能,測得其比電容值為303.6F/g(掃描速率50mV/s),阻抗為4.2Ω。附圖說明圖1:本發明實施例1制得的花狀MnO2納米片掃描電鏡照片。圖2:本發明實施例1制得的花狀MnO2納米片拉曼圖譜。圖3:本發明實施例1制得的花狀MnO2納米片的循環伏安曲線。具體實施方式以下結合附圖和具體實施例對本發明作具體的介紹。實施例1本實施例制備MnO2納米片電極材料的方法,步驟如下:S1、將0.1mol/L的過硫酸銨與0.1mol/L的硫酸錳在燒杯中混合溶解得到40mL的水溶液。S2、將燒杯放置在超聲恒溫水浴中,溫度為40℃,將10mL濃度為0.005mol/L的硫酸鎳溶液,勻速滴加至上述混合溶液中,滴加時間為3min。用保鮮膜封閉燒杯,反應1h。S3、將反應產物離心洗滌后-30℃冷凍干燥5h。實施例2制備步驟與實施例1相同,區別之處在于超聲恒溫水浴中,溫度為30℃,硫酸鎳的濃度為0.05mol/L。實施例3制備步驟與實施例1相同,區別之處在于超聲恒溫水浴中,溫度為60℃,硫酸鎳的濃度為0.1mol/L,反應時間為0.5h,將離心洗滌產物50℃烘干2h。實施例4制備步驟與實施例3相同,區別之處在于超聲恒溫水浴中,溫度為45℃,硫酸鎳的濃度為0.05mol/L,反應時間為2h。實施例5制備步驟與實施例3相同,區別之處在于超聲恒溫水浴中,溫度為45℃,硫酸鎳的濃度為0.005mol/L,反應時間為3h。性能測試將實施例1-5制得的電極材料與炭黑、聚四氟乙烯(粘合劑)按一定比例混合(無水乙醇做溶劑),在研缽中研磨后混合均勻,制成1cm×1cm大小的薄片狀。將泡沫鎳剪成1cm×5cm大小的矩形長條,把制備好的產物薄片壓在泡沫鎳的一端,制成測試電極。采用三電極體系測試其比電容值,制備的電極作為正極,對電極為2cm×2cm鉑片,參比電極為Ag/AgCl電極電極,電解質溶液為0.5mol/L的Na2SO4溶液,掃描速率為50mV/s,電壓窗口為0~0.5V,實施例1制得的MnO2電極的循環伏安曲線如圖2,實施例1-5比電容值如表1。表1實施例1-5制得樣品的比電容值樣品比電容值(F/g)實施例1303.6實施例2289.5實施例3274.6實施例4266.9實施例5257.6由圖1可知,本發明制備實施例1的MnO2納米片電極材料具有花狀,三維立體的結構不僅增加了材料的比表面積,還使得電極活性材料與電解質充分接觸,有利于電解質離子在電極體相中的擴散,顯著提升其電化學性能;圖2為實施例1制得MnO2納米片的拉曼圖譜,圖中642.9cm-1處出現拉曼信號峰,此處對應著錳氧八面體中的Mn-O鍵的對稱伸縮振動,此峰對應于α-MnO2的特征振動峰,說明此時錳氧化物的晶體結構為α-MnO2,同時該圖譜未顯示出其他峰位,表明摻雜離子主要起到引導MnO2生長的作用,未參與到反應中進而確保MnO2具有良好的晶體結構;其循環伏案曲線如圖3所示,并沒有觀察到明顯的氧化還原峰,且曲線接近于矩形,說明該材料在充放電過程中呈現為雙電層效應,較大的比表面積能夠確保其具有優異的電化學性能,同時對稱的循環曲線表明電極充放電可逆性優異,電極活性材料利用率較高,計算結果表明,其比電容值高達303.6F/g。綜上所述,本發明制備超電容用金屬離子摻雜花狀MnO2納米片電極材料的方法,制備流程短、效率高且成本低、環境污染小,制得的MnO2電極材料具有優異的電化學性能,且該方法反應體系簡單容易控制,具有良好的應用前景。以上顯示和描述了本發明的基本原理、主要特征和優點。本行業的技術人員應該了解,上述實施例不以任何形式限制本發明,凡采用等同替換或等效變換的方式所獲得的技術方案,均落在本發明的保護范圍內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1