一種高容量負極材料的制備方法與流程
本發明屬于能源技術領域,具體地,涉及一種高容量負極材料的制備方法。
背景技術:
隨著鋰離子電池向動力電池、儲能等領域的應用擴展,對鋰離子電池能量密度與功率密度提出越來越高的要求,特別是新能源汽車使用鋰離子電池,目前能量密度只能達到120~180Wh/kg,續航里程150公里,與行業標桿400公里續航里程的美國特斯拉新能源電動車相比,還有很大的差距。一直以來國家非常重視新能源汽車的發展,早在2012年工信部《節能與新能源汽車產業發展規劃(2012—2020)》提出“以純電驅動為新能源汽車發展和汽車工業轉型的主要戰略取向,到2015年純電動汽車和插電式混合動力汽車累計產銷量力爭達到50萬輛,純電動汽車和插電式混合動力汽車生產能力達200萬輛、累計產銷量超過500萬輛,到2020年動力電池模塊比能量達到300Wh/kg以上,成本降至1.5元/Wh以下”。
顯然,傳統的正負極材料已經無法滿足社會發展對高能量密度鋰離子電池的需求,迫切要求新型高能量密度的正負極材料推出。硅基、鍺基、錫基及氧化亞硅、氧化錫、氧化鐵等負極材料由于具有較高的理論容量,被認為是最有希望的下一代鋰離子電池負極材料,但是這些非碳基負極材料的導電性較差,而且有巨大的體積膨脹,容易形成不穩定的SEI膜,這三大問題一直制約這類非碳基負極材料的實際應用。
傳統的碳包覆方法通常是有機碳源在1000度以下直接進行裂解、碳化,在非碳基材料表面形成無定形碳,雖然導電性有所提升,但是非碳類負極材料體積膨脹很大,容易導致這層無定形碳破碎,影響SEI穩定性,從而使容量降低較快,制約了其實際應用。隨著石墨烯的快速發展,石墨烯與這類非碳基材料復合成為研究的熱點,石墨烯的高強度、高導電、柔軟性的特性完全可以承受這類非碳基負極材料巨大體積膨脹的影響。中國專利申請CN104934573A報道以糖類為碳源,在金屬催化劑作用下在納米硅顆粒表面形成石墨烯,增強納米硅的電化學性能,其中存在的缺點是由于石墨烯具有較高的比表面積,石墨烯復合納米硅負極材料也同樣具有較高的比表面積,SEI穩定性問題很難克服。
因此,有必要提供一種改進的高容量負極材料的制備方法克服以上問題。
技術實現要素:
為解決現有技術中存在的上述問題,本發明結合了石墨烯高導電性、高強度與硅高容量的特點,制備了硅表面原位構筑類石墨化碳層結構,形成類石墨化碳包覆的復合硅負極材料,具有突出的電化學性能,同時具有較低的成本,從而提供了一種改進的鋰離子電池高容量負極材料的制備方法。具體地說,本發明與現有技術的區別在于:本發明闡述在低溫下原位形成類石墨化碳提高非碳負極材料的導電性,并對非碳負極材料體積膨脹起到緩沖作用,與現有技術中形成的石墨烯有本質區別。本發明將無定形碳包覆易操作、可規模化生產、低成本等特點與石墨烯的高導電與高強度的特點相結合,在非碳負極材料表面制備了類石墨化包覆碳層,大大提高非碳負極材料的循環穩定性。
因此,本發明的目的在于提供一種高容量負極材料的制備方法。
為達到上述目的,本發明采用如下技術方案:
將可石墨化類有機碳源與催化劑及非碳類負極材料以干法或濕法混合在一起得到碳化前驅體,其中所述可石墨化類有機碳源、所述催化劑和所述非碳類負極材料的質量比為(0.03-0.8):(0-0.1):1,在惰性氣體保護狀態下,經過低溫200~400度碳化2~8h、高溫600~1200度碳化2~15h等過程,最后得到高容量負極材料。
其中,所述的可石墨化類有機碳源選自葡萄糖、石油瀝青、煤瀝青、聚丙烯腈、纖維素等可石墨化碳源中的至少一種。
所述的催化劑選自二價或三價鐵類、鎳類、鈷類及鎂氧化物或鹽中的至少一種。例如,氯化鐵、氯化亞鐵、硫酸鐵、硫酸亞鐵、硝酸鐵、硝酸亞鐵、二茂鐵、乙酸鐵、三氧化二鐵、硝酸鎳、硫酸鎳、氯化鎳、氯化鎂、硝酸鎂、醋酸鎂、氧化鈷、草酸鈷等中的至少一種。
所述的非碳類負極材料選自硅、鍺、錫等可鋰化的高容量金屬及其合金(包括硅鋁、硅鐵、硅鎳、硅錫、硅鍺、硅硼等)及氧化硅(SiOX,1≤x<2)、氧化錫、三氧化二鐵、氧化鈷、氧化鈦、氧化錳等中的至少一種,顆粒的尺寸范圍為0.01~20μm。
所述的干法是指將所述可石墨化類有機碳源、所述催化劑和所述非碳類負極材料混合,在200r/min條件下球磨0.5~10h,得到所述碳化前驅體。
所述的濕法是指將所述可石墨化類有機碳源、所述催化劑和所述非碳類負極材料分散于有機溶劑(乙醇、丙酮、二甲基甲酰胺、甲苯、四氫呋喃等)或水中,形成懸浮液,經過旋轉蒸發或噴霧干燥得到碳化前驅體。
有益效果:本發明涉及一種通過原位構筑的方法在高容量的非碳基類負極材料的表面原位構筑類石墨化碳層,形成類石墨化碳包覆的復合非碳負極材料,從而提高硅、錫等非碳基負極材料的導電性與循環穩定性,采用本發明制備的非碳基類負極材料具有高導電、高容量、高穩定性的電化學性能,并且具有較低的成本,能夠大規模用于高能量密度鋰離子電池,具有廣泛的工業應用前景。
附圖說明
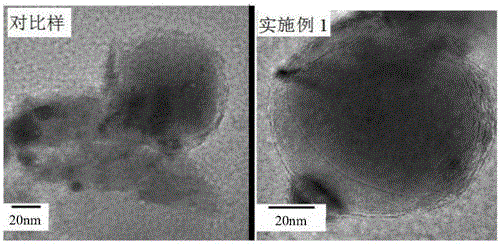
圖1為實施例1樣品的高分辨HRTEM照片。
圖2為拉曼譜圖,其中,圖2(a)為實施例1樣品,圖2(b)為實施例1對比樣品。
圖3為實施例1樣品與對比樣品的循環曲線。
圖4為實施例2樣品與對比樣品的循環曲線,其中,圖4(a)為實施例2樣品,圖4(b)為實施例2對比樣品。
圖5為實施例3樣品與對比樣品的循環曲線。
圖6為實施例4樣品與對比樣品的循環曲線。
圖7為實施例5樣品的循環曲線。
具體實施方式
以下結合具體實施例,對本發明做進一步說明。應理解,以下實施例僅用于說明本發明而非用于限制本發明的范圍。
除特別注明外,以下實施例中使用的原料均市售可得。
實施例1、樣品制備
取30~100nm硅粉10g,均勻分散到丙酮溶液中,加入3g石油瀝青,加入0.5g氯化亞鐵,在高速攪拌下混合均勻,用旋轉蒸發慢慢除去溶劑,將固體料取出,在惰性氣氛下,5度/分鐘升溫到300度,恒溫預碳化3小時,然后10度/分鐘升溫至850度,并恒溫8h,慢慢冷卻至室溫,取出固體,粉碎,用2M鹽酸除去樣品中殘留的鐵或鐵氧化物,去離子水洗滌,最后500度燒結2h后即得。
本實施例的對比樣品的區別僅在于不添加催化劑氯化亞鐵,其他原料配比及制備方法與實施例1樣品制備相同。
實施例2、樣品制備
取1μm氧化錫粉末10g,加入5g葡萄糖,加入1g硝酸鎳,在200r/min下進行球磨5h,混合均勻,然后在惰性氣氛下,5度/分鐘升溫到300度,恒溫預碳化5小時,然后10度/分鐘升溫至900度,并恒溫10h,慢慢冷卻至室溫,取出固體,并粉碎即得。
本實施例的對比樣品的區別僅在于不添加催化劑硝酸鎳,其他原料配比及制備方法與實施例2樣品制備相同。
實施例3、樣品制備
取50~100nm硅鍺合金粉末10g,加入3g煤瀝青,加入0.5g硝酸鐵與0.2g硝酸鎳,在200r/min下進行球磨5h,混合均勻,然后在惰性氣氛下,5度/分鐘升溫到400度,恒溫預碳化2小時,然后10度/分鐘升溫至900度,并恒溫6h,慢慢冷卻至室溫,取出固體,用6M的鹽酸除去金屬離子,真空200度烘干,粉碎即得。
本實施例的對比樣品的區別僅在于不添加催化劑硝酸鐵和硝酸鎳,其他原料配比及制備方法與實施例3樣品制備相同。
實施例4、樣品制備
取0.5~5μm氧化亞硅粉末10g,加入2g聚丙烯腈,加入0.5g硝酸鐵與0.2g硝酸鎂,溶于100ml二甲基甲酰胺中,高速攪拌3h,形成黏稠均勻的混合液,然后在惰性氣氛下,5度/分鐘升溫到260度,恒溫預碳化2小時,然后10度/分鐘升溫至850度,并恒溫6h,慢慢冷卻至室溫,取出固體,用6M的鹽酸除去金屬離子,真空200度烘干,粉碎即得。
本實施例的對比樣品的區別僅在于不添加催化劑硝酸鐵和硝酸鎂,其他原料配比及制備方法與實施例4樣品制備相同。
實施例5、樣品制備
取100~500nm硅鎳合金粉末10g(鎳含量20%),加入5g煤瀝青,在200r/min下進行球磨5h,混合均勻,然后在惰性氣氛下,5度/分鐘升溫到400度,恒溫預碳化2小時,然后10度/分鐘升溫至700度,并恒溫6h,慢慢冷卻至室溫,取出固體,粉碎即得。
由于本實施例的硅鎳合金中的鎳對碳層起到催化作用,不需要額外添加催化劑。
實施例6、TEM電鏡檢測
為了證明表面碳層的石墨化程度,我們選擇實施例1制備的樣品進行HRTEM電鏡檢測,結果如圖1所示,在2萬倍下的HRTEM照片,可以看到小于10nm的碳包覆層厚度,而且局部有規整的層狀排布,相應的對比樣品(沒有催化劑參與)HRTEM沒有觀察到明顯的層狀排布區。由于其他的實施例與實施例1有類似原理,因此不再一一表征。
實施例7、拉曼光譜檢測
將實施例1制備的樣品進行拉曼光譜檢測,結果如圖2所示。由圖2(a)可以看出,實施例1的樣品的拉曼譜圖中很明顯出現了G帶與D帶峰,G帶峰強度明顯高于D帶峰強度,說明有一定的石墨化程度。而相應的圖2(b)中對比樣品的拉曼譜圖中D帶峰強度明顯高于G帶峰強度,說明沒有催化劑條件下,硅表面包覆碳層完全是無定型形式。由于其他的實施例與實施例1反應條件類似,不再一一測試。
實施例8、扣式電池的性能評估
將實施例1~5中制備獲得的樣品進行扣式電池的性能評估,評估方法說明如下:
按照鋰離子電池負極電極制作工藝進行負極電極的制備。首先,按照一定質量比為0.8:0.10:0.10稱取活性材料、導電劑(Super P)、粘結劑(CMC與SBR),以水為溶劑。按照打膠、加導電劑、再加活性材料的順序加料,攪拌均勻后,用100um涂膜器將混好的電池漿料涂在10um的銅箔上,在100度烘箱中烘干水份,然后轉移到120度的真空烘箱中烘烤10h后,取出、輥壓(壓實密度按照1.0計算)至所需厚度,用扣式電池2025模具沖片,稱重、真空烘烤24h后,轉移至手套箱,裝配扣式電池、使用藍電充放電測試儀在0.1C倍率下進行循環測試,評價電池性能。
循環性能檢測結果如圖3-7所示。由圖3可以看出,實施例1的石墨化碳包覆的納米硅負極材料相對于對比樣品的循環穩定性明顯改善。同樣地,由圖4和圖5可以看出,實施例2和3的樣品相對于相應對比樣品的循環穩定性能明顯改善。由圖6可見,雖然實施例4制備獲得的樣品與對比樣品的循環穩定性相近,但是實施例4的樣品的容量明顯高于對比樣品。由圖7可見,實施例5制備獲得的樣品循環7~8周后,容量趨于穩定。
以上結果說明非碳類負極材料表面包覆碳層在催化劑作用下,轉化成類石墨化碳層,對于改善大體積膨脹非碳類負極材料的循環性能有非常明顯的效果。
綜上所述,本發明提供了一種制備類石墨化碳包覆非碳類負極材料的方法,類石墨化碳起到與石墨烯類似的功效,對非碳類負極材料在充放電過程中的體積膨脹起緩沖作用,有利于負極材料表面SEI膜穩定性,從而大大提高非碳類負極材料的循環穩定性。具體地說就是將可石墨化類有機碳源與非碳類負極材料及石墨化催化劑均勻混合,在一定溫度下進行碳化、石墨化,得到石墨化碳包覆的非碳類負極材料。在900℃以上,碳容易與硅、錫等金屬類非碳負極材料發生反應,因此通常碳包覆溫度低于900℃,這非常不利于碳的石墨化,催化劑的功能是降低石墨化溫度。石墨化碳層一方面具有較低的比表面積,降低反應過程中SEI的反復修復;另一方面具有較高的強度,對于大膨脹負極材料起到緩沖作用。采用本發明制備的非碳基類負極材料具有高導電、高容量、高穩定性的電化學性能,并且具有較低的成本,能夠大規模用于高能量密度鋰離子電池,具有廣泛的工業應用前景。
- 還沒有人留言評論。精彩留言會獲得點贊!