微量碳定量分析裝置和微量碳定量分析方法與流程
本發明涉及微量碳定量分析裝置和微量碳定量分析方法。
背景技術:
鋼是鐵和碳的合金,通過控制碳含量和加工熱處理方法而能夠分別制造具有各種特性的材料。因此,鋼鐵材料中的碳的定量分析是對控制材料特性極其重要的技術。
從這樣的觀點考慮,例如在開發用于分析鋼片和鋼水中的碳量的技術,對以質量%計為0.1%以下的碳的定量分析變得可能。例如在專利文獻1中記載了能夠以10ppm以下的精度推定鋼水中的碳濃度的方法。這些技術可對材料中作為平均組成的碳含量進行高精度分析。
另一方面,即便是相同的碳含量,也能夠通過加工熱處理方法而大幅改變鋼鐵材料的機械性質。決定該機械性質的是鋼鐵材料的金屬組織。在金屬組織的控制中,碳的μm級以下的分布和存在形態起到重要作用。因此,金屬組織的微量碳的分析是極其重要的。
在金屬組織的分析中經常使用光學顯微鏡和電子顯微鏡。特別是使用電子顯微鏡時,能夠觀察小于1μm的大小的金屬組織。而且,可以在電子顯微鏡中安裝對因入射電子束而從試樣產生的特性x射線進行檢測的機器。如果使用該機器,則能夠實現以碳為代表的、構成以小于1μm的大小分布的金屬組織的元素的定性、定量分析。
近年來,鋼鐵材料的組織控制不斷高度化,在同一材料內具有多個金屬組織的多相鋼的使用也不稀奇。因此,除了材料的平均化學組成以外,還對與金屬組織相應的局部的化學組成信息,尤其是對碳的分布進行分析變得越來越重要。
現有技術文獻
專利文獻
專利文獻1:日本特開2005-281750號公報
專利文獻2:日本專利第4499125號公報
技術實現要素:
為了以ppm級的高精度對鋼鐵材料中的碳進行定量,廣泛使用例如在專利文獻1中公開的濕式化學分析法。但是化學分析法是用于調查平均組成的,無法測定與金屬組織對應的局部的碳濃度。
如果使用在電子顯微鏡中安裝有特定的機器的分析電子顯微鏡,則能夠觀察金屬組織的同時進行局部區域的元素分析。作為該機器,可舉出使用了半導體檢測器的能量分散型x射線分光器(eds:energydispersivex-rayspectrometer)或使用了分光晶體和x射線檢測器的波長色散型x射線分光器(wds:wavelengthdispersivex-rayspectrometer)。特別是作為碳等輕元素的分析,通常利用靈敏度高的wds進行點分析或者進行邊掃描電子束邊分析的面分析。
然而,為了控制鋼鐵組織而含有的元素在很多情況下為0.01~1質量%以下的微量,因此后述的試樣污染(contamination)的影響成為問題。特別是碳為左右鋼鐵材料的特性的最重要的元素,污染的影響是極其重大的問題。
污染是指因電子束照射而微量存在于裝置真空中的有機物系的殘留氣體成分分解,屬于殘留氣體構成成分的碳和氫選擇性附著于試樣的電子束照射區域的現象,有時也表示附著物本身。
污染通過電子束照射而迅速形成并覆蓋試樣表面,因此除了觀察和分析變得困難以外,還因為污染的主成分為碳,所以在進行碳分析時,無法判定是試樣自身的碳還是分析了污染的碳。
因此,在使用電子顯微鏡的碳分析中,通常使用吸附試樣室內的殘留氣體而使成為污染源的殘留氣體量降低的液氮阱。
然而,液氮阱的效果也不充分,在微量碳的面分析中,污染隨著分析時間而增加,因而存在分析結束位置的碳分布的測定值比實際高的問題。
應予說明,在專利文獻2中公開了預先測定相對于分析測量時間的污染的碳的增加量并在定量時進行校正的方法。但是,污染的產生量有因測定條件所致的變動和日間變動,因此當測定時需要預先測定污染的增加量,不僅繁瑣,而且有時精度也不夠。
本發明是鑒于以上方面進行的,目的在于提供能夠沒有污染影響地分析試樣中的微量碳的微量碳定量分析裝置和微量碳定量分析方法。
本發明人等為了達成上述目的而進行了深入研究,結果發現通過并用液氮阱和等離子體或氧自由基發生裝置作為污染的抑制手段,并且設置2個以上的檢測碳的特性x射線的碳檢測部,從而能夠排除污染的影響的同時對試樣中的微量碳進行定量,進而完成了本發明。
即,本發明提供以下的(1)~(9)。
(1)一種微量碳定量分析裝置,具備:被抽真空的試樣室;通過液氮冷卻來捕獲所述試樣室內的殘留氣體的液氮阱;對所述試樣室內的試樣表面和所述試樣室內吹送等離子體或氧自由基的等離子體或氧自由基發生裝置;產生電子束的電子源;使所述產生的電子束會聚的透鏡部;在所述試樣室內的試樣上掃描所述會聚了的電子束或者在固定所述會聚了的電子束的狀態下驅動所述試樣室內的試樣的掃描驅動部;和檢測由照射了電子束的試樣發射的碳的特性x射線的碳檢測部;并且,設置2臺以上的所述碳檢測部,由所設置的2臺以上的所述碳檢測部檢測到的碳的特性x射線來對所述試樣室內的試樣中的微量碳進行定量。
(2)根據所述(1)所述的微量碳定量分析裝置,其中,進一步具備在電子束照射中將所述試樣室內的試樣加熱保持在50℃以上的試樣加熱部。
(3)根據所述(1)或(2)所述的微量碳定量分析裝置,其中,進一步具備對所述試樣室內的試樣表面進行濺射的濺射部。
(4)根據所述(1)~(3)中任一項所述的微量碳定量分析裝置,其中,進一步具備從外部環境放置試樣并被抽真空的試樣準備室和對所述試樣準備室內的試樣表面吹付等離子體或氧自由基的第2等離子體或氧自由基發生裝置。
(5)根據所述(1)~(4)中任一項所述的微量碳定量分析裝置,其中,所述碳檢測部為具備分光晶體和x射線檢測器的波長色散型x射線分光器。
(6)一種微量碳定量分析方法,使用了所述(1)所述的微量碳定量分析裝置,利用所述液氮阱來捕獲所述試樣室內的殘留氣體,在電子束照射前利用所述等離子體或氧自由基發生裝置對所述試樣室內的試樣表面和所述試樣室內吹送等離子體或氧自由基,由設有設置的2臺以上的所述碳檢測部檢測到的碳的特性x射線來對所述試樣室內的試樣中的微量碳進行定量。
(7)根據所述(6)所述的微量碳定量分析方法,其中,在電子束照射中將所述試樣室內的試樣加熱保持在50℃以上。
(8)根據所述(6)或(7)所述的微量碳定量分析方法,其中,在電子束照射前對所述試樣室內的試樣表面進行濺射。
(9)根據所述(6)~(8)中任一項所述的微量碳定量分析方法,其中,在對所述試樣室內的試樣進行面分析時,將比試樣的分析對象區域更向外側寬的區域作為照射電子束的電子束照射區域,僅對電子束照射所述分析對象區域的期間進行面分析。
根據本發明,能夠提供可不受污染的影響地分析試樣中的微量碳的微量碳定量分析裝置和微量碳定量分析方法。
附圖說明
圖1是表示微量碳定量分析裝置的一部分的結構示意圖。
圖2是表示微量碳定量分析裝置的結構示意圖。
圖3是表示并用液氮阱和等離子體發生裝置時與未并用兩者時的電子束照射時間與c-k特性x射線強度的關系的圖。
圖4是鋼中碳的面分析結果。
圖5是鋼中碳的線分析結果。
圖6是表示使用通常的試樣臺時和使用加熱臺時的電子束照射時間與c-k特性x射線強度的關系的圖。
圖7是表示面分析方法的一個例子的示意圖。
具體實施方式
首先,對完成本發明的經過進行說明。
作為在電子顯微鏡中的污染抑制的手段,眾所周知有液氮阱。本發明人等確認了在電子顯微鏡中搭載液氮阱則對污染的抑制有效。通過液氮冷卻實現了試樣中的碳濃度的濃淡的定性分析。但是,即便使用液氮阱,也無法避免分析中的污染的附著,因此停留于定性分析。
另外,本發明人等確認了通過使用對試樣和電子顯微鏡的試樣室內吹送等離子體或氧自由基的裝置同樣能夠進行碳的定性分析。但是,依然無法避免污染的附著。
接下來,本發明人等通過并用液氮阱和等離子體或氧自由基發生裝置(以下,也簡稱為“等離子體發生裝置”)來嘗試抑制污染。通過上述并用而進一步抑制了污染,但污染仍然附著,難以實現排除污染影響的碳分析。
然而,本發明人等對相對于分析時間的特性x射線的強度進行調查的結果發現隨著分析時間增加而污染堆積得較厚,碳的特性x射線的強度經時地增加。即,如果以短時間結束分析,則能夠實質上忽略污染的影響而進行碳分析。
可是在短時間的分析中分析的信號量不足,因此特別是在微量成分的分析中檢測本身變得困難。為了增加特性x射線的信號量而增加照射電流量是有效的。但是,與此相伴,來自基體的主成分的信號量也會變多。
在電子顯微鏡中通用地用于元素分析的eds能夠同時檢測所有元素的信號,但每單位時間能夠檢測和處理的信號量存在上限。因此使用eds時來自基體的信號量變得過量,所以為了分析微量成分而增加照射電流量也是有極限的。
因此,本發明人等著眼于屬于檢測特性x射線的另一手段的wds。wds無法同時檢測多種元素,但能夠排除來自基體的過量信號,僅分析目標元素,因此在微量成分分析時能夠增加電流量。
本發明人等使用搭載有wds的電子束顯微分析儀(epma:electronprobemicroanalyzer),并用液氮阱和等離子體發生裝置。其結果可知如果是點分析和線分析的話,通過使用碳濃度已知的試樣的標準曲線法,能夠以1質量%以下的精度對未知試樣的局部的碳濃度進行定量。
然而,1個點上的分析時間短的面分析中,雖然能夠捕捉定性碳濃度的濃淡,但因信號量少而無法進行定量分析。如果延長分析時間,則受污染的影響而無法進行正確的分析,因此本發明人等考慮提高x射線檢測器的靈敏度。
因為搭載于epma的wds一次僅測定一個元素,所以從便利性的觀點考慮,一般在epma中搭載多個wds。另外,可以想象到利用epma來分析多種多樣的試樣和元素,所以wds的分光晶體以能夠在一次分析中從輕元素到重元素進行測定的方式進行組合。而且,根據分析目的,需要區分使用波長分辨率高的分光晶體和能夠測定許多x射線強度的分光晶體而進行分析,因此在搭載于epma的wds中,一般盡可能組合不同類型的分光晶體。
這樣,以往為了對應多種多樣的試樣和分析目的而使搭載于epma的分光晶體的組合具有通用性。
與此相對,本發明人等嘗試將搭載于epma的多個wds的分光晶體中的至少2個以上置換成可檢測碳的特性x射線的分光晶體。其結果發現碳分析的靈敏度意想不到地提高,即便是1個點上的分析時間短的面分析,也能夠對其強度進行定量。
在具有復雜結構的金屬組織的分析中,與點分析和線分析相比,在面分析上得知試樣整體的元素濃度分布是極其重要的。然而,到目前為止并未發現利用面分析對碳進行定量分析的手段。
與此相對,本發明人等發現通過設為在epma中將可檢測碳的特性x射線的分光晶體和x射線檢測器各自設有至少2個以上的構成,能夠實現碳的定量面分析。還發現進一步通過并用液氮阱和等離子體發生裝置,其定量值成為不受污染影響的可靠性充分的值。用面分析進行微量碳的定量分析在以往的epma中沒有,是全新的功能。另外,用面分析進行微量碳的定量分析對于以鋼鐵為代表的材料開發而言是極其重要且劃時代的技術。
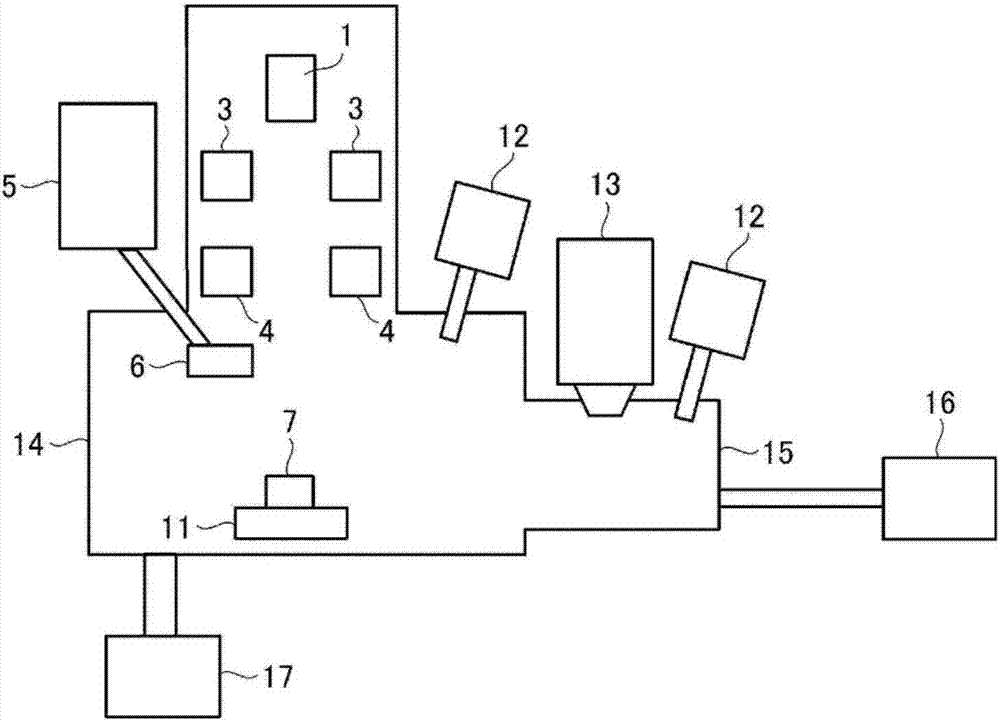
接下來,基于圖1和圖2,對本發明的微量碳定量分析裝置(以下,也簡稱為“裝置”)的一個實施方式進行說明。
圖1是表示微量碳定量分析裝置的一部分的結構示意圖。圖2是表示微量碳定量分析裝置的結構示意圖。
由電子源1產生的電子束2因作為透鏡部的會聚透鏡3和物鏡4會聚。會聚的電子束2因作為掃描驅動部的掃描線圈a(參照圖7)對配置于試樣臺11的試樣7上進行掃描。或者,在會聚的電子束2固定的原樣下由作為掃描驅動部的試樣臺11來驅動試樣7。這樣,對試樣7照射電子束2。因電子束2的照射而從試樣7產生的(發射的)特性x射線8由分光晶體9導入到x射線檢測器10中,記錄來自試樣7的特性x射線的強度。應予說明,關于裝置構成參照圖7。
分光晶體9和x射線檢測器10在裝置中各自安裝有至少2個以上。而且,所安裝的多個分光晶體9中的至少2個以上為可分光碳的特性x射線的元件。作為這樣的元件,例如可舉出人工多層膜lde(layereddispersionelement)。
由作為可分光碳的特性x射線的元件的分光晶體9和x射線檢測器10構成的、檢測碳的特性x射線的碳檢測部至少為2個以上,更優選3個以上。上限沒有特別限定,根據試樣室14的空間制約而適當地設定。
僅進行碳分析時,可以使全部的分光晶體9和x射線檢測器10為可分光碳的構成。在需要分析碳以外的元素時,可以使分光晶體9中的幾個設為可分析這些元素的構成。
液氮阱6被由液氮罐5補充的液氮冷卻,吸附試樣室14的殘留氣體,抑制污染的產生。
如上所述,試樣7在試樣室14內由試樣臺11驅動。應予說明,作為試樣臺11,可以使用后述的加熱臺。
在試樣室14中設置等離子體或氧自由基發生裝置12,使其在對試樣7照射電子束2之前或之后運轉。等離子體或氧自由基不僅使試樣7的表面清潔,還有抑制由試樣室14產生的污染的效果,因此優選等離子體或氧自由基發生裝置12設置在試樣室14。
另外,除了試樣室14以外,也可以隔著開關自如的隔壁(未圖示)與試樣室14鄰接配置的試樣準備室15中設置等離子體或氧自由基發生裝置12(第2等離子體或氧自由基發生裝置)。
此時,首先,以關閉隔壁的狀態從外部環境向試樣準備室15中放置試樣7,一邊用真空泵16抽真空,一邊使用試樣準備室15的等離子體或氧自由基發生裝置12對試樣準備室15內的試樣7的表面吹送等離子體或氧自由基。接下來,打開隔壁而使試樣7移動到試樣室14之后,使用試樣室14的等離子體或氧自由基發生裝置12,對試樣室14內的試樣7的表面和試樣室14內吹送等離子體或氧自由基。
應予說明,在沒有試樣準備室15地以抽出方式將試樣7直接放置到試樣室14中的裝置中,僅在試樣室14中設置等離子體或氧自由基發生裝置12。
因試樣7的前調整和電子束2的照射而厚厚地堆積在試樣7的表面的污染有時利用等離子體或氧自由基發生裝置12也難以除去。為了在裝置內將其除去,可以搭載用于對試樣7的表面進行濺射的濺射部的離子槍13。應予說明,有時因ar濺射等的離子濺射而使周圍污染,因此優選離子槍13不在試樣室14中,而設置在試樣準備室15中。另外,離子濺射不僅具有除去試樣7的表面的污染而露出清潔的試樣7的表面從而進行準確的碳分析的效果,還有除去試樣7的表面的研磨應變相,在金屬材料時在試樣表面形成與微細組織對應的微細凹凸的效果。由此使用裝置的顯微鏡功能來尋找感興趣的測定對象變得容易。
試樣室14和試樣準備室15分別由真空泵17和真空泵16來抽真空。
然而,裝置內的污染源除了附著于試樣的污染以外,還有由裝置自身產生的污染,例如有時也由作為裝置的必需構成的真空泵帶入污染源。該污染源是由真空泵內部的油引起的,因此優選使用不用油的真空泵。應予說明,在對向大氣開放的試樣準備室進行粗抽時經常使用的旋轉泵中,有時從其逆流內部的油成分。
因此,作為試樣準備室15的真空泵16和試樣室14的粗抽用真空泵(未圖示),例如優選使用干泵。另外,作為用于設成更高真空的試樣室14的真空泵17,例如優選使用渦輪分子泵,而并非油擴散泵。
接下來,基于圖3,對并用液氮阱和等離子體或氧自由基發生裝置的技術性意義進行說明。
本發明人等將并用液氮阱和等離子體發生裝置時的污染的附著狀態與未并用它們的情況進行了對比。更詳細而言,使用日本電子公司制的epma(jxa-8530f)測定了對epma用鐵-碳標準試樣(c量:0.089質量%,剩余部分:鐵)照射電子束時產生的c-k特性x射線強度。對在試樣的相同位置照射電子束時的累積的電子束照射時間與測定的x射線強度的關系進行了調查。
圖3是表示并用液氮阱和等離子體發生裝置時與未并用兩者時的電子束照射時間與c-k特性x射線強度的關系的圖。
如圖3的坐標圖所示,在比較例(不使用液氮阱和等離子體發生裝置)中,隨著電子束照射時間增加,c-k特性x射線強度也上升,由此可知在試樣表面以c為主成分的污染在緩慢堆積。
與此相對,在僅使用液氮阱時(不使用等離子體發生裝置),相對于照射時間,x射線強度幾乎恒定,可知抑制了新的污染的堆積。
接下來,在上述epma的試樣室中安裝ibssgroup公司制的等離子體發生裝置(gv10xds),產生等離子體5分鐘,使試樣表面和試樣室清潔化。接著,使用液氮阱進行同樣的測定的結果,與僅使用液氮阱的情況相比,c-k特性x射線強度進一步降低。由此可知因等離子體發生裝置除去了試樣表面的污染,能夠測定與試樣本來的c含量對應的x射線強度。另外,如果與僅使用液氮阱時相比,x射線強度存在差別,因此可知僅使用液氮阱時也測定了原本附著于表面的污染。試樣表面的污染根據試樣制備的正時而附著量改變,因此在僅使用液氮阱時無法進行具有反復的再現性的碳分析。通過并用利用液氮阱進行的新污染的堆積的抑制和利用等離子體發生裝置進行的表面附著污染的除去,從而首次能夠正確地測定了試樣中原本含有的碳。
然而,從圖3的坐標圖可知,如果電子束照射時間超過50秒,則即便并用液氮阱和等離子體發生裝置時,雖然是極少但確認到了隨著電子束照射時間的增大而x射線強度上升。圖3是對試樣的相同位置進行分析的結果,因此如果向電子束未照射區域改變分析位置,則還能夠實現沒有污染影響的分析。
然而,在使用epma的線分析和面分析中,因為連續地掃描電子束而進行分析或者連續地驅動試樣而進行分析,所以在之前的分析位置的污染的影響會累積。即便并用液氮阱和等離子體發生裝置時,在分析時間超過30分鐘的面分析(映射分析,mappinganalysis)中,分析結束之前c-k特性x射線強度上升,因污染而無法測定正確的碳分布。通過在點分析和線分析中并用液氮阱和等離子體發生裝置,能夠在出現污染的影響之前結束測定,但在測定時間長的面分析中,兼得足夠的信號量的確保和污染的抑制的測定在以往的epma中難以實現。
接下來,基于圖4,對設置2個以上的檢測碳的特性x射線的碳檢測部的技術性意義進行說明。
如上所述,通過并用液氮阱和等離子體或氧自由基發生裝置來掃描電子束而進行面分析或者驅動試樣而進行面分析時,從電子束照射開始到30分鐘能夠抑制污染影響地進行分析。將分析時間設為30分鐘時,例如以256×256像素進行面分析則像素一個點上約成為25m秒的分析時間。在該分析時間下的碳的定量精度由x射線量的統計分布程度(statisticaldispersion)估計約為0.5質量%(3σ)。在鋼的分析中,該精度不夠,僅能作為碳量大小的定性的差異。
此時,通過增加檢測碳的特性x射線的碳檢測部(分光晶體和x射線檢測器)能夠提高靈敏度。例如,通過同時使用3個碳檢測部(分光晶體和x射線檢測器)來測量c-k特性x射線量,能夠將分析精度提高到0.3質量%。在鋼的面分析中,該定量分析精度的差別極其大。
圖4是鋼中碳的面分析結果。試樣是具有fe-0.13c-1.4si-2mn的組成(數字為質量%)的鐵素體-馬氏體雙相鋼。比較例和發明例都對通過改變熱處理溫度而使鐵素體分率變化的兩鋼種進行了分析。已知鐵素體相中的碳量為0.02質量%以下,大多添加碳分布在作為鐵素體相以外的第二相的馬氏體相。因此,即便是相同的碳添加量,也因鐵素體分率的差異而使馬氏體相中的碳濃度不同。
應予說明,在圖4中,碳濃度由濃淡(明暗)表示,例如,鐵素體相相當于顏色深的(暗的)區域,馬氏體相相當于顏色淺的(明亮的)區域。
圖4的比較例是并用液氮阱和等離子體發生裝置,使用日本電子公司制的epma(jxa-8530f),僅使用1個檢測c-k特性x射線的碳檢測部(分光晶體和x射線檢測器)而進行碳的面分析的例子。分光晶體為人工多層膜lde6h。以分析像素數256×256且在每一點像素以20m秒進行面分析。如圖4所示,在比較例中,能夠確認鐵素體相與馬氏體相的碳濃度差,但信號量少,因此馬氏體相中的濃淡(明暗)沒有清晰地顯現,無法確認在鐵素體分率不同的兩鋼種間的馬氏體相中的碳濃度的差別。
另一方面,圖4的實施例是用3個碳檢測部同時檢測c-k特性x射線時的分析例。分光晶體為人工多層膜lde6h。除了鐵素體相與馬氏體相的碳濃度差以外,馬氏體相中的濃淡(明暗)也清晰地顯現,能夠確認在鐵素體分率不同的兩鋼種間的馬氏體相中的碳濃度的差異。
本發明通過基于并用液氮阱和等離子體或氧自由基發生裝置而產生的污染的大幅減少以及基于同時使用2個以上的碳檢測部而產生的對微量碳的靈敏度的提高,從而使碳的定量分析值的可靠性大幅提高。
因此,與現有技術相比,不僅在如基于圖4所說明的面分析中,在點分析和線分析中都具有優越性。
圖5是鋼中碳的線分析結果。試樣為具有fe-0.15c-2.0si-1.5mn的組成(數字為質量%)的鐵素體馬氏體多相鋼。
在圖5的比較例中,進行基于液氮阱的污染的減少的同時僅用1個碳檢測部進行線分析,但隨著分析位置變大而鐵素體相的碳定量值(圖5中,由虛線的箭頭表示)上升。這并非反映實際的濃度變化,而是在分析中堆積的污染使定量值上升的結果,碳濃度0.05質量%以下的定量值無法信賴。
另一方面,在圖5的發明例中,并用液氮阱和等離子體發生裝置,并且用3個碳檢測部進行線分析,即便分析位置變大,也沒有看到碳定量值(圖5中,由粗線的箭頭表示)的上升,能夠沒有污染影響地對僅試樣中本來含有的碳進行正確的定量。
關于碳濃度比鐵素體相高的馬氏體相的碳定量值,在圖5的發明例與比較例之間也能看到差別。試樣的馬氏體相中的碳濃度在基于擴散控制型相變計算軟件的理論計算中為0.32質量%。另一方面,圖5的發明例中平均為0.33質量%,圖5的比較例中為0.38質量%。即,在發明例中,能夠相對于理論上的預測值以0.01質量%的誤差進行分析,但在比較例中,污染的分析誤差為0.05質量%以上,而且,在分析的起點和終點無法避免因污染所致的定量值的變動。
接下來,基于圖6,對使用加熱臺(試樣加熱部)作為裝置的試樣臺(圖2中,由符號11表示)的情況進行說明。通過使用能夠將試樣加熱保持在50℃以上的加熱臺,能夠延長從開始電子束照射到污染在試樣表面堆積為止的時間,進一步減少污染的影響。
本發明人等對使用正溫度系數(ptc:positivetemperaturecoefficient)熱敏電阻,將試樣加熱保持在110℃而進行點分析時的電子束照射時間與c-k特性x射線強度的關系進行了調查。更詳細而言,以電子束照射時間為10秒時的x射線強度為基準,調查了相對于電子束照射時間的從該基準強度的差。使用日本電子公司制的epma(jxa-8530f),設有3個檢測碳的特性x射線的碳檢測部。并用液氮阱和等離子體發生裝置,在開始照射電子束進行分析之前用試樣準備室的離子槍對試樣表面進行ar濺射。將結果示于圖6。
圖6是表示使用通常的試樣臺時和使用加熱臺時的電子束照射時間與c-k特性x射線強度的關系的圖。
如圖6的圖所示,使用通常的試樣臺時,如果電子束照射時間超過50秒,則x射線強度顯著上升,即便并用液氮阱和等離子體發生裝置,也看到了污染的影響。另一方面,使用加熱臺而進行試樣的加熱保持時,即便電子束照射時間超過100秒,x射線強度也不上升,能夠進一步減少污染的影響。
因此,在面分析中也能夠延長直到污染附著于試樣的時間,能夠實現定量精度更高的分析。
應予說明,加熱溫度小于50℃時,防止污染的附著的效果小。另一方面,如果加熱溫度過高,則有時發生試樣的變質,另外,也無法抑制因熱所致的試樣漂移,因此加熱溫度優選150℃以下。
另外,本發明人等對附著于試樣表面的污染進行了調查。其結果發現污染并沒有均勻地堆積在試樣表面的電子束照射區域,而是尤其集中堆積在電子束照射區域的邊緣。即,污染附著在電子束照射區域的邊緣而無法實現正確的碳量的定量,但電子束照射區域的中央部能夠實現碳的定量分析。因此,在對試樣進行面分析時,如果使試樣中的比目標分析對象的區域(分析對象區域)更向外側寬的區域作為照射電子束的區域(電子束照射區域),僅在電子束照射該分析對象區域的期間進行面分析,則能夠去除電子束照射區域的邊緣,能夠實現減少污染影響的分析。
圖7是表示面分析方法的一個例子的示意圖。電子束2通過掃描線圈a對試樣中的電子束照射區域c的內部進行掃描。在控制裝置e中輸入基于掃描線圈a的電子束2的位置信息和x射線強度信號。控制裝置e僅在電子束2掃描分析對象區域b的期間向圖像輸出裝置f輸出x射線強度信號。基于x射線強度信號,通過與標準試樣數據強度的比較,從而能夠進行定量分析。
應予說明,在分析對象區域b和電子束照射區域c都為四方形的情況下,分析對象區域b的外框與電子束照射區域c的外框之間的距離d在分析對象區域b的一邊的長度為10μm以下時優選為1μm~10μm,更優選2μm~7μm。另外,在分析對象區域b的一邊的長度大于10μm時,距離d相對于分析對象區域b的一邊的長度優選0.1~1.0倍,更優選0.2~0.7倍。
符號說明
1:電子源
2:電子束
3:會聚透鏡(透鏡部)
4:物鏡(透鏡部)
5:液氮罐
6:液氮阱
7:試樣
8:特性x射線
9:分光晶體(碳檢測部)
10:x射線檢測器(碳檢測部)
11:試樣臺(掃描驅動部,試樣加熱部)
12:等離子體或氧自由基發生裝置
13:離子槍(濺射部)
14:試樣室
15:試樣準備室
16:真空泵
17:真空泵
a:掃描線圈(掃描驅動部)
b:分析對象區域
c:電子束照射區域
d:距離
e:控制裝置
f:圖像輸出裝置
- 還沒有人留言評論。精彩留言會獲得點贊!