一種硫化氫分離裝置及檢測浸膏香料中硫化氫含量的方法與流程
本發明屬于分析化學技術領域,具體是涉及一種能夠有效分離煙用浸膏類香料中硫化氫的裝置。同時,本發明還涉及一種準確測定煙用浸膏類香料中硫化氫含量的方法。
背景技術:
煙用香精和料液是卷煙生產中不可或缺的重要原料,隨著煙草工業的發展,各種天然香料,如:煙草提取物、植物提取物、水果提取物、花類提取物、微生物發酵產物、海藻提取物等被廣泛應用于卷煙工業中。天然提取物的使用彌補了合成類香料成分簡單、香味單一的不足,香氣成分更加豐富飽滿,可為卷煙產品賦予合成香料無法比擬的特殊香韻。
浸膏類香料,作為最重要的天然香料之一,是通過有機溶劑浸提天然植物或動物含香分泌物后,經過濾、濃縮后得到的一種膏狀物,具有高水分、高糖的基質特點。在浸提過程中,植物或動物原料中的蛋白質、氨基酸等也容易進入到浸膏中,從而為微生物提供了良好的生長繁殖條件,使得浸膏類香料在生產、加工及儲運過程中均存在腐敗變質的風險。腐敗變質的香料一旦不慎流入到卷煙產品中,將給卷煙產品的品質和風格特征造成無法挽回的負面影響。當浸膏類香料發生腐敗變質時,其中的蛋白質、氨基酸等會分解產生硫化氫,伴隨有臭雞蛋氣味。因此,硫化氫的含量可作為浸膏類香料是否發生腐敗變質的檢測指標。
現有技術中,硫化氫含量的檢測方法主要有亞甲基藍法、碘量法、醋酸鉛反應速率法、紫外分光光度法等,但這些方法靈敏度較低,僅適用于常規分析;而且這些方法大都操作步驟冗長費時,需使用多種化學試劑,易受到共存物的干擾而影響分析結果的準確性。新近出現的離子色譜法由于具有檢測快速、靈敏度高、選擇性好、可同時測定多組分等優點而受到廣泛關注,尤其是它可以用來檢測通過其他方法難以測定的離子,例如陰離子等。用離子色譜法測定硫化氫雖已有文獻記載,但將其用于煙用浸膏類香料中硫化氫含量的測定還未見相關報道。
技術實現要素:
本發明的目的在于針對現有技術的不足,提供一種基于水蒸汽蒸餾法、能夠快速有效地分離煙用浸膏類香料中硫化氫的裝置。
本發明的目的還在于提供一種檢測煙用浸膏類香料中硫化氫含量的方法。先蒸餾分離出煙用浸膏類香料中的硫化氫,然后運用離子色譜法準確測定硫化氫的含量,進而根據浸膏香料樣品與標準品的硫化氫含量的差值就可以對樣品的腐敗變質情況作出判定。
本發明的目的通過以下技術方案予以實現。
除非另有說明,本發明所采用的百分數均為質量百分數。
一種有效分離煙用浸膏類香料中硫化氫的裝置,其特征在于:包括蒸餾燒瓶(1)、接收器(2)和吸收液貯存瓶(8);所述的接收器(2)的主體部分呈倒u形,右側為蒸餾進樣通道,左側為冷凝液收集通道(10);接收器(2)的頂部設置有封閉冷凝管(3),蒸餾進樣通道、冷凝液收集通道(10)和封閉冷凝管(3)相互連通;所述的蒸餾進樣通道的外側設置有加酸漏斗(4),蒸餾進樣通道的底部與蒸餾燒瓶(1)氣密連通;所述的冷凝液收集通道(10)的底端伸入吸收液貯存瓶(8);所述的吸收液貯存瓶(8)的頂部設置有排氣口(7),底部設置有放樣閥門(9)。
優選的,所述的接收器(2)與封閉冷凝管(3)通過磨口氣密連通;所述的蒸餾進樣通道與蒸餾燒瓶(1)通過磨口氣密連通。
所述的加酸漏斗(4)頂部設有磨口,用于放置磨口玻璃塞(5),底部設有開關閥門(6)。
所述的冷凝液收集通道(10)的內徑為0.5-1cm,冷凝液收集通道(10)的底端距離吸收液貯存瓶(8)底部0.5-1.5cm。
所述的排氣口(7)的口徑為0.2-0.5cm。
所述的吸收液貯存瓶(8)的外壁有計量刻度,加酸漏斗(4)的外壁有計量刻度。
所述的有效分離煙用浸膏類香料中硫化氫的裝置采用石英玻璃燒制。
本發明分離裝置的工作過程:稱取煙用浸膏類香料樣品置于蒸餾燒瓶1中,加入超純水混勻;從排氣口7向吸收液貯存瓶8中加入硫化氫吸收液;打開加酸漏斗4的開關閥門6,定量酸液流入蒸餾燒瓶1中,然后關閉開關閥門6。加熱蒸餾燒瓶1使樣品水溶液沸騰,樣品中的硫化氫隨水蒸氣一并餾出,經封閉冷凝管3冷凝后由冷凝液收集通道進入吸收液貯存瓶8。蒸餾完畢后打開放樣閥門9,放出吸收液,然后再從排氣口7加入純水洗滌吸收液貯存瓶8,合并洗滌液和吸收液用于分析檢測。
一種準確測定煙用浸膏類香料中硫化氫含量的方法,包括以下步驟:
(1)稱取2.0~10.0g煙用浸膏類香料樣品置于250ml的蒸餾燒瓶中,加入50ml超純水混勻,然后加入磷酸調節溶液的ph值≤2,得到樣品水溶液;
(2)在吸收液貯存瓶中加入10ml5%氫氧化鈉溶液,備用;
(3)將樣品水溶液加熱至160℃,控制蒸餾速度為2~4ml/min,樣品中的硫化氫隨水蒸氣一并餾出,水蒸氣冷凝后進入吸收液貯存瓶,被氫氧化鈉溶液吸收;當蒸出餾分達到25~35ml時停止蒸餾,收集吸收液;再用5.0~8.0ml的純水洗滌吸收液貯存瓶,合并洗滌液和吸收液,并用純水準確定容到50ml;
(4)采用脈沖安培檢測法測定合并溶液中的硫化氫含量:
其中,采用ionpacas7離子色譜柱;離子色譜流動相為:1.5mol/l氫氧化鈉:1.0mol/l乙酸鈉:2%體積分數乙二胺,體積比40:50:10;柱溫:30℃;流速:1.0ml/min;進樣量:10μl;檢測器的檢測電位波形為:0min(-100mv)、0.2min(-100mv)、0.9min(-100mv)、0.91min(-1000mv)、0.93min(-300mv)、1.0min(-300mv);
將硫離子標準儲備液用1%的氫氧化鈉溶液逐級稀釋成10μg/ml、2.5μg/ml、0.8μg/ml、0.2μg/ml、0.08μg/ml和0.025μg/ml的系列標準溶液,線性回歸分析,通過濃度和對應的峰面積進行回歸得到工作曲線;再對合并溶液進樣分析,根據其峰面積從工作曲線計算出硫化氫的濃度,結合樣品處理流程,得到樣品中硫化氫的含量值。
基于上述方法,根據樣品中硫化氫的含量與標準品中硫化氫含量的差值就可以進一步判斷煙用浸膏類香料的品質狀態。一般來說,當樣品中硫化氫的含量超過浸膏標準品硫化氫含量的50%時,說明樣品已發生明顯變質。
與現有技術相比,本發明具有如下優點:
1、本發明的裝置結構簡單,連接部件和接口相比于傳統的水蒸氣蒸餾裝置大大減少,安裝、蒸餾操作更方便簡單,成本更低,氣密性更佳。其中,加酸漏斗可在密閉的條件下加入酸,有效避免了樣品前處理過程中硫化氫的損失,有利于檢測工作的精確度與檢測數據的穩定性。同時,吸收液可通過貯存瓶的排氣口加入,蒸餾完全后可從放樣閥門排出合并溶液直接供儀器分析用;當處理完一個樣品后,通過更換蒸餾燒瓶,并且從排氣口加水清洗接收器后即可處理下一個樣品,極大的節省了檢測時間,提高了檢測效率。
2、蒸餾分離后的硫化氫采用脈沖安培檢測法測定,檢測靈敏度遠高于常用的滴定法和分光光度法。而且通過離子交換柱,硫離子可和樣品中其它共存的干擾成分完全分離,避免了光度法和滴定法中因共存干擾成分影響而有可能產生假陽性的缺陷。該方法操作簡單,檢測快速,靈敏度高、選擇性好、精密度高。
3、運用該方法的檢測結果還能夠進一步準確判定煙用浸膏的變質情況,并作出相應的風險預警,降低產品變質的風險,對保障煙用浸膏類香精和料液的質量安全具有重要的現實意義。
附圖說明
圖1為本發明分離裝置的結構示意圖;
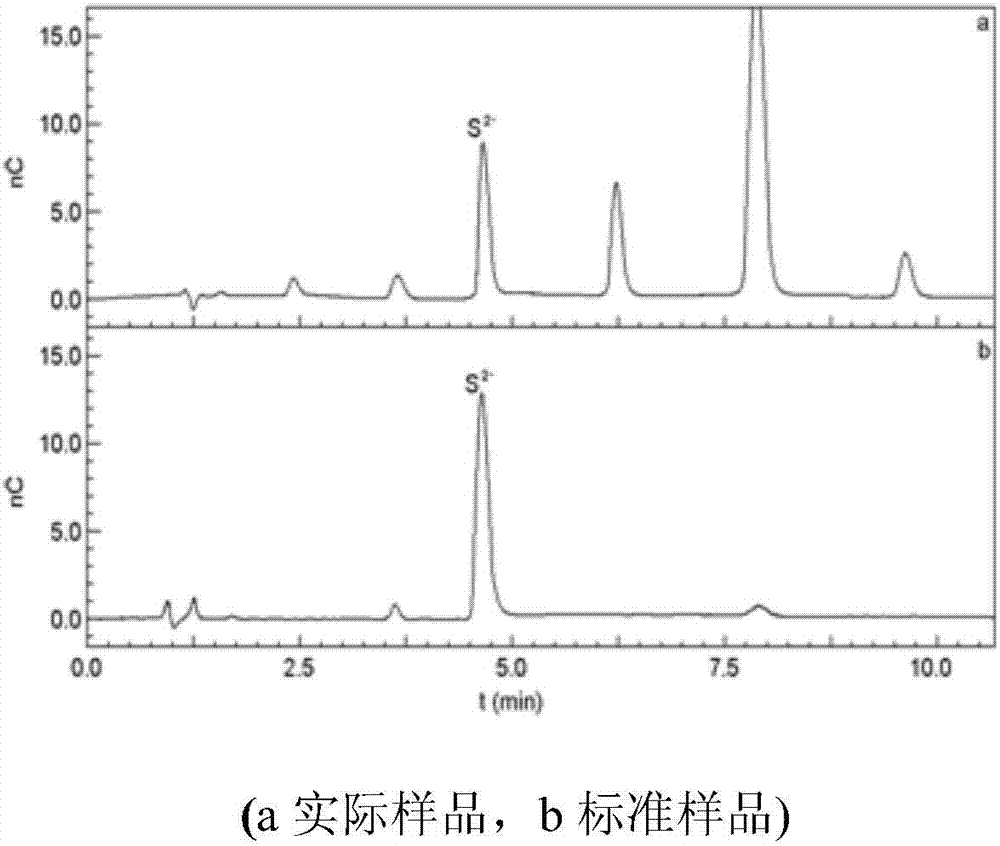
圖2為浸膏香料實際樣品和標準樣品的離子色譜圖。
具體實施方式
以下結合附圖和實施例對本發明作進一步的詳細說明,但附圖和實施例并不是對本發明技術方案的限定,所有基于本發明教導所作出的變化或等同替換均勻屬于本發明的保護范圍。
裝置實施例1
如圖1所示,一種有效分離煙用浸膏類香料中硫化氫的裝置,各部件均采用石英玻璃燒制而成。
具體包括蒸餾燒瓶(1)、接收器(2)和吸收液貯存瓶(8),所述的接收器(2)的主體部分呈倒u形,右側為蒸餾進樣通道,左側為冷凝液收集通道(10);在接收器(2)的頂部設置有封閉冷凝管(3),接收器(2)與封閉冷凝管(3)通過磨口氣密連通;蒸餾進樣通道、冷凝液收集通道(10)和封閉冷凝管(3)相互連通。在所述的蒸餾進樣通道的外側設置有加酸漏斗(4),加酸漏斗(4)的底部通過管道與蒸餾進樣通道連通,管道上設有開關閥門(6),加酸漏斗(4)頂部設有磨口,用于放置磨口玻璃塞(5)。蒸餾進樣通道的底部與蒸餾燒瓶(1)通過磨口氣密連通。所述的冷凝液收集通道(10)的底端伸入吸收液貯存瓶(8),所述的吸收液貯存瓶(8)的頂部設置有排氣口(7),底部設置有放樣閥門(9)。
所述的冷凝液收集通道(10)的內徑為0.5-1cm,冷凝液收集通道(10)的底端距離吸收液貯存瓶(8)底部0.5-1.5cm。所述的排氣口(7)的口徑為0.2-0.5cm。
所述的吸收液貯存瓶(8)的外壁有計量刻度,加酸漏斗(4)的外壁有計量刻度。
該分離裝置的工作過程:稱取煙用浸膏類香料樣品置于蒸餾燒瓶1中,加入超純水混勻;從排氣口7向吸收液貯存瓶8中加入硫化氫吸收液;打開加酸漏斗4的開關閥門6,定量酸液流入蒸餾燒瓶1中,然后關閉開關閥門6。加熱蒸餾燒瓶1使樣品水溶液沸騰,樣品中的硫化氫隨水蒸氣一并餾出,經封閉冷凝管3冷凝后由冷凝液收集通道進入吸收液貯存瓶8。蒸餾完畢后打開放樣閥門9,放出吸收液,然后再從排氣口7加入純水洗滌吸收液貯存瓶8,合并洗滌液和吸收液用于硫化氫含量的分析檢測。
本發明檢測方法的工藝條件優化及思路
1、蒸餾條件
煙用浸膏類香料中的硫化氫可在酸性條件下蒸餾出,因此需加入酸以調節樣品水溶液的ph值。常見的可供選擇的酸有:硫酸、磷酸、硝酸和鹽酸等,其中硝酸和鹽酸的揮發性太強,且沸點過低,故不適合在本發明中使用;硫酸和磷酸的沸點都比較高,蒸餾時不易揮發,但硫酸具有強腐蝕性,操作安全性不如磷酸。因此,在本發明中選用磷酸作為ph值的調節試劑。實驗結果表明,當在蒸餾燒瓶中加入10ml(1+1)磷酸水溶液(磷酸和水的體積比為1:1)時,樣品水溶液的ph值可控制在2以下,足夠使樣品中的硫化氫完全餾出。
蒸餾出的硫化氫采用堿性溶液吸收,氫氧化鈉溶液由于具有較好的吸收效果,且在氫氧化鈉溶液中硫化氫以游離態硫離子形態存在,很適合后續的離子色譜分析。因此,本發明選取氫氧化鈉溶液作為硫化氫的吸收液。實驗結果表明,采用10ml5%的氫氧化鈉溶液可完全吸收蒸餾出的硫化氫。
蒸餾溫度是影響硫化氫蒸出速度的關鍵因素,隨著溫度的升高,蒸出速度會明顯加快。但如果蒸餾速度過快,會導致硫化氫的吸收效率降低;而蒸餾速度過慢又會耗費更長的時間。實驗結果表明,餾出液速度應控制在2~5ml/min,此時硫化氫的回收率基本穩定。設置電熱套的加熱溫度為160℃,蒸餾速度為3ml/min左右。當蒸餾出的餾分達到25ml以上時,樣品中的硫化氫已完全蒸出(在殘余液中繼續加入水再蒸餾已沒有硫離子檢出)。因此,本發明選取收集25~35ml的餾分。
2、離子色譜條件的選擇
離子色譜中常用的檢測方法主要有電導檢測法、熒光檢測法、紫外-可見光分光光度檢測法與安培檢測法。其中,安培檢測法是一種電化學檢測技術,是用于測量電化學活性物質在工作電極表面發生氧化還原反應時產生電流變化的檢測器,具有結構簡單、靈敏度高、選擇性好及響應范圍寬等優點。根據施加電位方式的不同,安培檢測法又可以分為脈沖安培檢測法、恒電位安培檢測法和積分脈沖安培檢測法。由于s2-在電極上的氧化反應產物會毒化電極表面,影響檢測靈敏度和基線平穩。為解決上述問題,本工作采用脈沖安培檢測法,在工作電位后再施加清洗電位,可使靈敏度重新恢復,避免了直流檢測的靈敏度會隨電極表面的活性下降而降低的缺陷。
ionpacas7離子色譜柱為疏水型氫氧化物選擇型陰離子交換柱,可分離多種多價陰離子,包括多聚磷酸鹽、多聚膦酸鹽、多價絡合劑,和六價鉻等金屬陰離子。采用該離子交換柱分離硫離子出峰時間短、色譜峰峰型好、能和樣品中其它共存成分完全分離,因此選擇該色譜柱。對應的淋洗液為:1.5mol/l氫氧化鈉:1mol/l乙酸鈉:2%(體積分數)乙二胺(40:50:10)。在該條件下,樣品中的硫離子均能得到很好的分離,且分離時間短。隨著進樣量的增加,分析靈敏度提高,但色譜峰會稍變寬。本實驗中,當進樣量為10μl時靈敏度已完全能滿足實際樣品的分析需求,因此選擇進樣量為10μl。在選定實驗條件下,標樣和實際浸膏樣品的色譜圖見附圖-2。
3、工作曲線與檢測限
對硫離子標準儲備液逐級稀釋,配制一系列的標準工作溶液,在選定的檢測條件下進樣50μl進行分析。根據檢測器測得的峰面積a對硫化氫的含量c(單位:μg/ml)進行線性回歸分析,得到線性方程:a=33.84c–2.675,相關系數r=0.9996,離子色譜峰面積與濃度呈現較好的線性關系,線性范圍為0.05~10μg/ml,檢測限為3.2ng/ml,定量限為12ng/ml。
4、精密度
為了考察本方法的精密度,我們對五種不同類型的變質煙用浸膏樣品進行了7次日內重復測定。實驗結果顯示,硫化氫的相對標準偏差在3.0%~3.4%之間,表明該方法的日內精密度較好。同時,我們也對此五種不同類型的變質煙用浸膏樣品進行了7天的重復性實驗,硫化氫的相對標準偏差在3.3%~3.8%之間,表明該方法的日間精密度較好。
表1方法精確度試驗結果
tab.1precisionofthemethodin5realsample(n=6)
5、加標回收率
本方法選取了五種不同類型的模擬霉變煙用香精和料液,分別進行了高(50.0μg)、中(20.0μg)、低(8.0μg)三個水平的加標回收率實驗,硫化氫的加標回收率在92.0%~95.4%之間,表明該方法檢測精準、可靠。
表2硫化氫的加標回收率試驗結果
tab.2recoveriesoftheh2sspikedinarealsample(n=6)
6、硫化氫的測定和煙用浸膏變質的判定
根據煙用浸膏樣品的變質情況,酌情準確稱取2.0~10.0g于250ml蒸餾燒瓶中,加入50ml超純水。待按圖1所示搭好蒸餾裝置后,從排氣口加入10ml5%氫氧化鈉溶液作為硫化氫吸收液。接著打開加酸漏斗的閥門,加入10ml(1+1)的磷酸溶液,關閉閥門,啟動電熱套加熱升溫至160℃。隨著反應溫度的升高,蒸餾燒瓶中的樣品水溶液逐漸開始沸騰,硫化氫隨水蒸氣一并餾出,經冷凝后被氫氧化鈉溶液吸收。當蒸出餾分達到25~35ml時停止蒸餾,打開放樣閥門,放出吸收液;并從排氣口加入5.0~8.0ml的水洗滌吸收瓶,合并洗滌液并準確定容到50ml,供離子色譜分析用。
離子色譜流動相為:1.5mol/l氫氧化鈉-1.0mol/l乙酸鈉-2%(體積分數)乙二胺(40:50:10,體積比);柱溫:30℃;流速:1.0ml/min;進樣量:10μl;檢測器(工作電極:ag,參比電極:ag/agcl)的檢測電位波形為:0min(-100mv)、0.2min(-100mv)、0.9min(-100mv)、0.91min(-1000mv)、0.93min(-300mv)、1.0min(-300mv)。通過實際樣品中硫化氫的峰面積,從工作曲線計算硫化氫的含量,并通過硫化氫含量變化判定煙用浸膏的變質情況。判定標準為樣品中硫化氫的增加超過樣品硫化氫背景值的50%時,樣品已存在發生變質的風險。
方法實施例1
測定樣品為紅棗浸膏,準確稱取4.0g于250ml蒸餾燒瓶中,加入50ml超純水。按圖1所示搭好蒸餾裝置后,從排氣口加入10ml5%氫氧化鈉溶液作為硫化氫吸收液。接著打開加酸漏斗的閥門,加入10ml(1+1)的磷酸溶液,關閉閥門,啟動電熱套加熱升溫至160℃。隨著反應溫度的升高,蒸餾燒瓶中的樣品水溶液逐漸開始沸騰,硫化氫隨水蒸氣一并餾出,經冷凝后被氫氧化鈉溶液吸收。當蒸出餾分達到30ml時停止蒸餾,打開放樣閥門,放出吸收液;并從排氣口加入5.0~8.0ml的水洗滌吸收瓶,合并洗滌液并用純水準確定容到50ml,供離子色譜分析用。
離子色譜流動相為:1.5mol/l氫氧化鈉-1.0mol/l乙酸鈉-2%(體積分數)乙二胺(40:50:10,體積比);柱溫:30℃;流速:1.0ml/min;進樣量:10μl;檢測器(工作電極:ag,參比電極:ag/agcl)的檢測電位波形為:0min(-100mv)、0.2min(-100mv)、0.9min(-100mv)、0.91min(-1000mv)、0.93min(-300mv)、1.0min(-300mv)。
硫離子標準儲備液(cas:lzry,0.1mg/ml,購自上海超研生物科技有限公司),將該標準儲備液用1%的氫氧化鈉逐級稀釋成10μg/ml、2.5μg/ml、0.8μg/ml、0.2μg/ml、0.08μg/ml、0.025μg/ml的系列標準溶液,線性回歸分析,通過濃度和對應的峰面積進行回歸得到工作曲線,實際樣品進樣分析后,通過其峰面積從工作曲線計算硫化氫的濃度,結合樣品處理流程即可得到樣品中硫化氫的含量。
本實施例中,通過實際樣品中硫化氫的峰面積,從工作曲線計算樣品中的硫化氫含量為88.6μg/g,而相同工藝新制備的紅棗浸膏對照樣中的硫化氫含量為16.5μg/g,說明樣品已發生了明顯的腐敗變質。
方法實施例2
測定樣品為無花果浸膏,準確稱取2.5g于250ml蒸餾燒瓶中,加入50ml超純水。按圖1所示搭好蒸餾裝置后,從排氣口加入10ml5%氫氧化鈉溶液作為硫化氫吸收液。接著打開加酸漏斗的閥門,加入10ml(1+1)的磷酸溶液,關閉閥門,啟動電熱套加熱升溫至160℃。隨著反應溫度的升高,蒸餾燒瓶中的樣品水溶液逐漸開始沸騰,硫化氫隨水蒸氣一并餾出,經冷凝后被氫氧化鈉溶液吸收。當蒸出餾分達到25ml時停止蒸餾,打開放樣閥門,放出吸收液;并從排氣口加入5.0~8.0ml的水洗滌吸收瓶,合并洗滌液并準確定容到50ml,供離子色譜分析用。
離子色譜流動相為:1.5mol/l氫氧化鈉-1.0mol/l乙酸鈉-2%(體積分數)乙二胺(40:50:10,體積比);柱溫:30℃;流速:1.0ml/min;進樣量:10μl;檢測器(工作電極:ag,參比電極:ag/agcl)的檢測電位波形為:0min(-100mv)、0.2min(-100mv)、0.9min(-100mv)、0.91min(-1000mv)、0.93min(-300mv)、1.0min(-300mv)。通過實際樣品中硫化氫的峰面積,從工作曲線計算樣品中的硫化氫含量為98.4μg/g,而相同工藝新制備的無花果浸膏對照樣中的硫化氫含量為10.62μg/g。說明樣品已發生了明顯的腐敗變質。
方法實施例3
測定樣品為煙草浸膏,準確稱取8.0g于250ml蒸餾燒瓶中,加入50ml超純水。按圖1所示搭好蒸餾裝置后,從排氣口加入10ml5%氫氧化鈉溶液作為硫化氫吸收液。接著打開加酸漏斗的閥門,加入10ml(1+1)的磷酸溶液,關閉閥門,啟動電熱套加熱升溫至160℃。隨著反應溫度的升高,蒸餾燒瓶中的樣品水溶液逐漸開始沸騰,硫化氫隨水蒸氣一并餾出,經冷凝后被氫氧化鈉溶液吸收。當蒸出餾分達到35ml時停止蒸餾,打開放樣閥門,放出吸收液;并從排氣口加入5.0~8.0ml的水洗滌吸收瓶,合并洗滌液并準確定容到50ml,供離子色譜分析用。
離子色譜流動相為:1.5mol/l氫氧化鈉-1.0mol/l乙酸鈉-2%(體積分數)乙二胺(40:50:10,體積比);柱溫:30℃;流速:1.0ml/min;進樣量:10μl;檢測器(工作電極:ag,參比電極:ag/agcl)的檢測電位波形為:0min(-100mv)、0.2min(-100mv)、0.9min(-100mv)、0.91min(-1000mv)、0.93min(-300mv)、1.0min(-300mv)。通過實際樣品中硫化氫的峰面積,從工作曲線計算樣品中的硫化氫含量為35.6μg/g,而相同工藝新制備的煙草浸膏對照樣中的硫化氫含量為6.76μg/g。說明樣品已發生了明顯的腐敗變質。
方法實施例4
測定樣品為沉香浸膏,準確稱取10.0g于250ml蒸餾燒瓶中,加入50ml超純水。待按圖1所示搭好蒸餾裝置后,從排氣口加入10ml5%氫氧化鈉溶液作為硫化氫吸收液。接著打開加酸漏斗的閥門,加入10ml(1+1)的磷酸溶液,關閉閥門,啟動電熱套加熱升溫至160℃。隨著反應溫度的升高,蒸餾燒瓶中的樣品水溶液逐漸開始沸騰,硫化氫隨水蒸氣一并餾出,經冷凝后被氫氧化鈉溶液吸收。當蒸出餾分達到30ml時停止蒸餾,打開放樣閥門,放出吸收液;并從排氣口加入5.0~8.0ml的水洗滌吸收瓶,合并洗滌液并準確定容到50ml,供離子色譜分析用。
離子色譜流動相為:1.5mol/l氫氧化鈉-1.0mol/l乙酸鈉-2%(體積分數)乙二胺(40:50:10,體積比);柱溫:30℃;流速:1.0ml/min;進樣量:10μl;檢測器(工作電極:ag,參比電極:ag/agcl)的檢測電位波形為:0min(-100mv)、0.2min(-100mv)、0.9min(-100mv)、0.91min(-1000mv)、0.93min(-300mv)、1.0min(-300mv)。通過實際樣品中硫化氫的峰面積,從工作曲線計算樣品中的硫化氫含量為6.47μg/g,而相同工藝新制備的沉香浸膏對照樣中的硫化氫含量為7.26μg/g。樣品中的硫化氫含量未發生明顯變化,說明樣品已未發生腐敗變質。
綜上所述,本發明的蒸餾分離裝置與現有的蒸餾裝置相比,連接部件和接口大大減少,使裝置的安裝、蒸餾操作更方便簡單、快速,成本更低,而且由于連接口減少,整套蒸餾裝置的氣密性更佳。蒸餾分離后的硫化氫采用離子色譜法測定,檢測靈敏度遠高于常用的滴定法和分光光度法。而且通過離子交換柱,硫離子可和樣品中其它共存的干擾成分完全分離,避免了光度法和滴定法中因共存干擾成分影響而有可能產生假陽性的缺陷。該方法操作簡單,檢測快速,靈敏度高、選擇性好、精密度高。運用該方法的檢測結果還能夠進一步準確判定煙用浸膏的變質情況,并作出相應的風險預警,降低產品變質的風險,對保障煙用香精和料液質量安全具有重要的現實意義。
- 還沒有人留言評論。精彩留言會獲得點贊!