一種湖泊沉積物中單酯磷組分的提取及磷核磁共振譜圖分析方法與流程
本發明屬于環境化學分析測試方法技術領域,具體涉及一種湖泊沉積物中單酯磷組分的提取及磷核磁共振譜圖分析方法。
背景技術:
磷是水生生態系統關鍵的限制性營養元素之一,過量的磷導致藻類大量生長繁殖,引起水質惡化并危害生態系統安全。近年來,包括生活污水、工農業生產、土壤徑流等過程攜帶的外源磷輸入得到了一定的控制,湖泊內源磷的釋放成為引起湖泊水質進一步惡化的關鍵因素。沉積物釋放是湖泊內源磷污染重要的來源之一,而有機磷是湖泊沉積物中磷的重要組成部分。已有研究表明,有機磷(po)在水生生態系統的磷循環中扮演重要角色,一些po組分可通過酶水解等過程轉化為生物可利用性活性磷酸鹽,進而成為富營養化湖泊藍藻水華持續暴發的重要磷源。
當前,沉積物中有機磷分析方法包括磷31核磁共振技術(31p-nmr)、酶水解技術、基于同步輻射光源x射線吸收精細結構分析技術、液相色譜或離子色譜與電感耦合等離子發射光譜連用(hplc-icp-aes)及液相色譜或離子色譜與電子噴霧離子化質譜聯用技術(hplc-esi-ms/ms)等。worsfold等人在2008年發表的文章《characterisationandquantificationoforganicphosphorusandorganicnitrogencomponentsinaquaticsystems:areview》(analyticachimicaacta.624(1):37-58(2008))中詳細介紹了各種分析檢測技術。近年來,31p-nmr作為定性定量分析手段在湖泊沉積物樣品的磷組分研究中得到廣泛運用,獲得了一些沉積物磷組分的基本信息,如正磷酸鹽、磷酸單酯、磷酸二酯、焦磷酸鹽、多聚磷酸鹽、磷脂和膦酸鹽等在沉積物中的含量及部分結構組成信息。
但是,湖泊沉積物提取液31p-nmr譜圖單酯磷區域中含有大量不同結構的有機磷化合物,如肌醇六磷酸鹽同分異構體包括肌-肌醇六磷酸鹽、鯊-肌醇六磷酸鹽和新-肌醇六磷酸鹽,葡萄糖磷酸鹽以及腺苷單磷酸鹽等。這些單酯磷在液相31p-nmr譜圖中的化學位移基本介于3.0ppm至6.5ppm小范圍之內(以正磷酸鹽化學位移定為6.0ppm為基礎)。一方面,沉積物中有機磷含量較低;另一方面,這些有機磷峰受提取液中ph及順磁性金屬離子(如fe3+等)影響,各峰之間相互重疊,峰展寬。這導致了沉積物提取液31p-nmr譜圖中單酯磷區域的分辨率不高,甚至對譜圖中單酯磷化合物的錯誤解析。尤其是我國幅員遼闊,不同湖泊沉積物基本性質差異很大,不同沉積物樣品提取液中背景金屬離子等含量差異很大,導致磷化學位移存在差異,尤其是31p-nmr譜圖中單酯磷具體組分的判斷造成了很大的困難或誤判。
為了消除磁性離子對31p核磁共振分析精確度的干擾,現有技術中已有相關報道,如中國專利《用31p核磁共振分析環境樣品時消除磁性離子干擾的方法》即公開了一種采用8-羥基喹啉沉淀去除含磷提取液中大量的鐵、錳等磁性離子,從而大幅度降低31p-nmr波譜峰寬,顯著提高磷組分的鑒別及定量分析的方法。該方法消除磁性離子干擾的效果顯著,且在處理過程中不造成磷組分的損失,操作簡便、成本低。
又如中國專利《一種鈣質沉積物有機磷的提取及組成分析方法》即公開了一種先采用偏酸性的乙二胺四乙酸溶液對沉積物進行沖洗,絡合去除部分鐵錳等順磁性金屬離子,再結合傳統方法使用naoh+edta將沉積物中的有機磷提取出來,通過31p-nmr進一步分析提取液中有機磷的組成。該方法適用于金屬元素ca/(fe+al)>0.7的鈣質沉積物。
雖然上述分析方法可一定程度上消除順磁性金屬離子對總有機磷31p核磁共振分析的干擾,但并未公開采用31p核磁共振分析單酯磷的具體方法。而且,一方面由于湖泊沉積物在采用堿液(naoh、edta或其組合)連續長時間提取后獲得,而這個長時間提取過程會造成一些不穩定單酯磷的降解;另一方面,一些不穩定的二酯磷也容易大量降解為單酯磷,所以,現在提取和前處理技術對于結合31p-nmr精確分析沉積物中單酯磷具有局限性。
更為關鍵的是,現有技術中,對湖泊沉積物提取液31p-nmr譜圖中單酯磷的解析主要采用以下方法:(1)以正磷酸鹽化學位移為參考,調節單酯磷化學位移,參考已知文獻報道各單酯磷化學位移,對各單酯磷進行定性和定量分析;(2)提取液中直接加入磷標準物質,如亞甲基二磷酸鹽(mdpa,methylenediphosphonicacidsodium),以該磷標準物質化學位移為參考,調節單酯磷化學位移并參考文獻報道,對各單酯磷進行定性和定量分析;(3)與沉積物提取同步,設定一個加入有機磷標準樣品的背景溶液,同步處理后,分析其中有機磷標準化合物化學位移,以此為參考并結合文獻報道,判斷沉積物提取液中各單酯磷組分。然而,實踐證明,這些分析方法存在以下問題:(1)對比發現,以正磷酸鹽化學位移為參考,不同文獻報道31p-nmr譜圖磷化合物化學位移均存在差異。尤其是單酯磷,由于化學位移集中且相互靠近,以正磷酸鹽化學位移為參考,參照不同文獻報道的化學位移會得出不同的分析結果;(2)提取液中直接加入磷標準物質,為不影響單酯磷的判別,往往采用遠離單酯磷化學位移區域的標準物質,如mdpa,其化學位移為17-18ppm之間,遠離單酯磷區域,以此為標準,參照文獻報道,也無法準確判斷31p-nmr譜圖中相應化學位移對應的單酯磷化合物;(3)沉積物中提取液的離子濃度等均會影響磷化合物化學位移,與沉積物提取同步,設定一個加入有機磷標準樣品的背景溶液獲得的模擬標準樣品,其離子濃度與有機質等均與沉積物提取液相差很大,實踐證明,二者的化學位移差異很大,尤其是單酯磷區域。
另外,湖泊類型多種多樣,不同區域湖泊地質背景差異很大,沉積物基本性質如礦物組成等差異大,導致沉積物有機磷提取液中離子強度、有機質含量等背景值差異大,這直接導致不同沉積物提取液中31p-nmr譜圖化學位移差異。因此,以上傳統的分析方法中仍沒有可靠的方法得到湖泊沉積物提取液31p-nmr譜圖中單酯磷組分的準確的分析結果。
技術實現要素:
本發明的目的是提供一種湖泊沉積物中單酯磷組分的提取及磷核磁共振譜圖的分析方法,一方面,該方法可以消除順磁性金屬離子對湖泊沉積物中單酯磷組分的干擾,還能最大程度地避免由于有機磷自身的不穩定性引起的降解;另一方面,本申請提供的分析方法避免了單酯磷峰相互重疊及其不穩定性引起的分析結果誤差。
為了實現上述目的,本發明提供了一種湖泊沉積物中單酯磷組分的提取及磷核磁共振譜圖分析方法,其包括如下步驟:
步驟一,用na2s溶液預處理湖泊沉積物提取濃縮液,得待測液1;
步驟二,對待測液1進行31p-nmr譜圖分析;
其中,步驟二的具體操作如下:
(1)測得待測液1的31p-nmr譜圖a;
(2)配制50mmol/l的單酯磷標準化合物、二酯磷標準化合物和膦酸鹽標準化合物溶液,將其加入步驟1中的待測液1后測得31p-nmr譜圖b;
(3)調節31p-nmr譜圖a和b的正磷酸鹽位移至6.00ppm,比對譜圖a和b,確定沉積物中相同物質的峰2-3個,參照31p-nmr譜圖b,調節31p-nmr譜圖a的化學位移;
(4)基于譜圖b中的單酯磷標準化合物化學位移,確定樣品譜圖a中單酯磷組分;
(5)利用單酯磷的峰面積占總面積的比例及提取液中總磷的含量,計算各單酯磷的含量。
優選地,步驟一中所述的na2s溶液的濃度為0.5-2mol/l;進一步優選為0.8-1.2mol/l;更進一步優選為1mol/l。
其中,所述na2s應相對過量,一方面可沉淀除去溶液中的fe3+、mn2+等順磁性離子,另一方面可使可能剩余的fe3+還原為fe2+,使沉積物提取濃縮液維持還原環境,還可以除去沉積物中的al3+、ca2+、mg2+,降低溶液鹽度,可更好地消除金屬離子對31p-nmr分析樣品時的干擾。與現有技術中的其它沉淀劑,如8-羥基喹啉相比,不僅省去了加入大量酸中和堿的過程,簡化了操作;而且,避免了強酸性物質(8-羥基喹啉溶液由12mol/l濃hcl配制而成)的引入導致的不穩定單酯磷和二酯磷的降解。
進一步優選地,步驟一中待測液1的制備方法具體如下:
向湖泊沉積物提取濃縮液中加入na2s的d2o溶液進行金屬離子的沉淀及還原處理,處理后的濃縮液于4℃條件下,靜置沉淀15-20h后,2000-10000g條件下,離心分離20-30min,得上清液,即為待測液1。
優選地,所述湖泊沉積物提取濃縮液是采用以下方法提取并濃縮得到:
1)采用naoh與edta的混合溶液多次、短時間和長時間結合連續提取湖泊沉積物樣品后合并提取液,所述多次即提取次數不少于3次,短時間即提取時間大于0小于等于4h,長時間即提取時間大于4小于等于16h;
2)測定提取液中的總磷,將提取液濃縮、干燥后加d2o溶解,得湖泊沉積物提取濃縮液。
進一步優選地,步驟1)中naoh與edta的混合溶液的濃度為naoh0.2-0.5mol/l,edta30-55mmol/l;更進一步優選為naoh0.2-0.4mol/l,edta40-55mmol/l,如naoh0.25mol/l,edta50mmol/l。
進一步優選地,步驟1)中使用naoh與edta的混合溶液提取湖泊沉積物樣品的次數為3-5次,更進一步優選為3次;每次提取時間逐漸增加,如提取時間依次分別為0.5-2h,2-5h,10-16h;如依次分別為2h,4h,16h。采用多次、短時間和長時間結合、時間逐漸增加的方法連續提取湖泊沉積物樣品可以避免因單酯磷及多酯磷的降解引起的31p核磁共振譜圖中單酯磷組分分析結果誤差。
提取次數小于3次時,要充分提取出樣品中的磷,需延長每次提取時間,但由于提取時間過長,提取過程中部分單酯磷、多酯磷降解,導致磷核磁共振譜圖組分分析得到的結果誤差增大;
提取次數太多,如超過5次時,提取液體積大,濃縮后鹽都太高,提取得到的有機磷和單酯磷的再溶解以及后續31p-nmr分析均受到影響,最后的分析結果誤差也會顯著增加。
進一步優選地,提取時的溫度為4-25℃。
進一步優選地,步驟2)中測定提取液中的總磷是采用磷鉬藍分光光度法或者采用電感耦合等離子體發射光譜儀(icp-oes)分析測定。
優選地,步驟(2)中單酯磷標準化合物至少包括肌醇六磷酸鹽myo-ihp、葡萄糖6磷酸鹽g6p、葡萄糖1磷酸鹽g1p、α-甘油磷酸鹽α-gly、β-甘油磷酸鹽β-gly、單磷酸腺苷amp、膽堿磷酸pcho;二酯磷化合物至少包括dna-p;
膦酸鹽標準化合物至少包括亞甲基二磷酸鹽mdpa。
優選地,步驟(1)中待測液1的核磁共振儀的條件設置為:溫度為25℃,脈沖為45℃,脈沖持續時間為5.3μs,采集時間為0.6s,馳豫時間設置為5s,掃描次數為12000~15000次。
優選地,步驟(3)的操作步驟為:首先,調節31p-nmr譜圖a和b的正磷酸鹽位移至6.00ppm;然后,調節譜圖a和b中化學位移范圍為6.5ppm至1.5ppm的信號峰高度至可對譜圖a和b中單酯磷區域峰信號進行精細對比;最后,確定沉積物中相同物質的峰2-3個,對比相同物質的峰位移,參照譜圖b,調節譜圖a中該峰化學位移至一致。
對于步驟(4),當樣品中出現標準樣品以外的譜峰時,以標準樣品化學位移為參考,通過參考已公開文獻和相對單酯磷標準化合物標準單酯磷化學位移,分析其它單酯磷的組分,其中單酯磷組分包括:肌醇磷酸鹽異構體myo-ihp(inositolhexakisphosphate)、chiro-ihp、neo-ihp和scyllo-ihp;chiro-ihp包括chiro-ihp(2e/4a)和chiro-ihp(4e/2a);d-肌醇1,4,5-三磷酸鹽d-myo-itp(d-myo-inositol-1,4,5-triphosphate);α-甘油磷酸鹽α-gly(glycerophosphate,β-甘油磷酸鹽β-gly(glycerophosphate;葡萄糖磷酸鹽g6p(glucose-6-phosphate)、g1p(glucose-1-phosphate);單核苷酸nuc(mononucleotides);磷酸膽堿pcho(cholinephosphate)。
與現有技術相比,本申請提供的湖泊沉積物中單酯磷組分提取及其磷核磁共振譜圖分析方法具有如下優點:
(1)本申請在提取過程中加入過量的na2s溶液,可在提取液堿性條件下消除順磁性金屬離子對31p-nmr分析樣品時的干擾的同時避免對不穩定性單酯磷和二酯磷的影響,保證了單酯磷組分分析的精確性。
(2)由實施例內容可以得知,本申請采取加標準化合物溶液對比的分析方法,可精確分析得出11種單酯磷組分及其對應含量,而采用上述傳統方法至多只能分析得出5-8種單酯組分,甚至會因對化學位移判斷錯誤而獲得錯誤的解析結果。另外,本申請得到的單酯磷組分總含量比傳統分析方法高出3%-10%。
(3)本發明的優選方案提取濃縮液的方法可優先提取出穩定性較差的有機磷,尤其是單酯磷,磷提取率明顯提高。其中,對沉積物總磷的提取率是傳統的一次長時間提取方法(只提取一次,提取時間16h以上)提取率的1-1.5倍;沉積物單酯磷的提取率是傳統的一次長時間提取方法提取率的1-2倍,對于湖泊沉積物而言,其提取效果明顯優于傳統提取工藝。
附圖說明
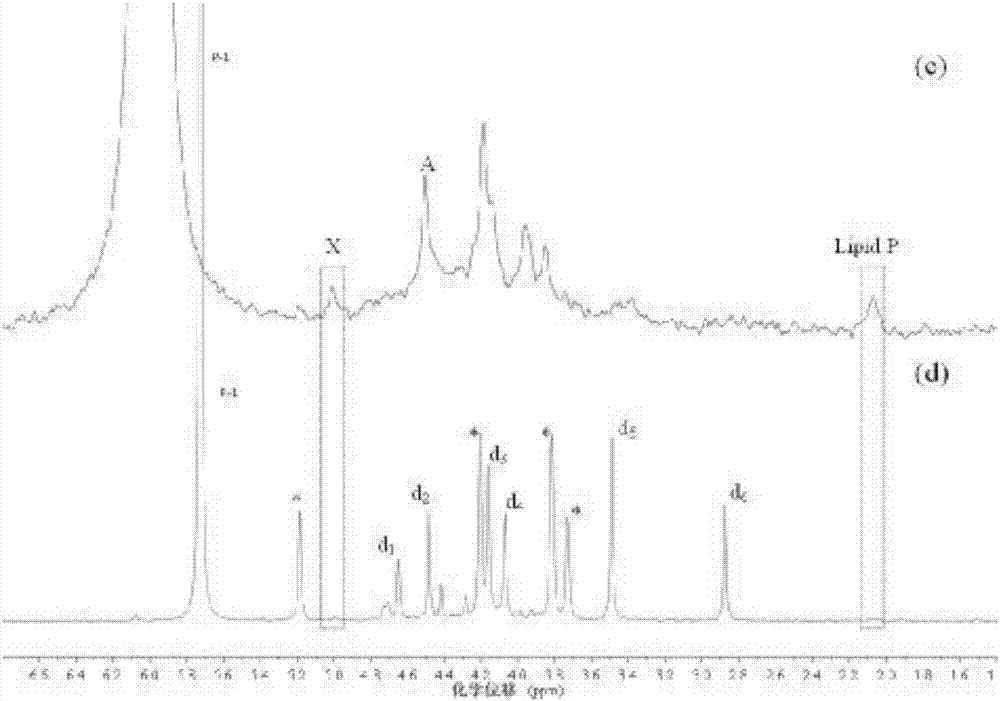
圖1實施例1中的dc-1沉積物樣品naoh-edta提取液以及加入磷標準化合物31p-nmr譜圖單酯磷區域對比
圖2實施例1中的dc-1沉積物樣品naoh-edta提取液以及加入磷標準化合物31p-nmr譜圖調節后單酯磷區域對比
注:①圖-1、2中加入的單酯磷標準化合物磷標準物質包括肌醇六磷酸鹽(myo-ihp);d1,葡萄糖6磷酸鹽(g6p);d2,α-甘油磷酸鹽(α-gly);d3,β-甘油磷酸鹽(β-gly);d4,5’單磷酸腺苷(amp);d5,膽堿磷酸(pcho);d6,葡萄糖1磷酸鹽(g1p);
②圖-1、2中(a)和(c)為沉積物naoh-edta提取液31p-nmr譜圖;(b)和(d)為對應提取液加入單酯磷標準化合物后31p-nmr譜圖。
③圖1中31p-nmr譜圖對比以正磷酸鹽為參考,將其位移調節為6.00ppm;圖2中31p-nmr譜圖以加入單酯磷標準化合物后的譜圖為參照,調節沉積物naoh-edta提取液31p-nmr譜圖。
具體實施方式
下面以我國滇池湖泊沉積物樣品提取液的磷核磁圖譜中單酯磷組分分析為例,對本方法作進一步說明。這些實施例僅是出于解釋說明的目的,而不限制本發明的范圍和實質。
本發明的所需試劑:
1)堿性提取劑:稱取10gnaoh和18.6gedta(na鹽)溶解到1.0l去離子水,配置成0.25mnaoh+50mmedta混合液。
2)na2s溶液:稱取480mgna2s·9h2o,加2mld2o溶解,配制成1m的na2s/d2o溶液。
3)k2s2o8溶液:將5g過硫酸鉀(k2s2o8)溶解于去離子水,并稀釋至100ml,配制成50g/l的溶液。其中,采用k2s2o8溶液消解的方法按照gb11893-89中規定的方法執行。
4)磷標準化合物溶液:稱取磷標準化合物(包括亞甲基二磷酸鹽(mdpa)、肌-肌醇六磷酸鹽(myo-ihp)、葡萄糖6磷酸鹽(g6p)、葡萄糖1磷酸鹽(g1p)、α-甘油磷酸鹽(α-gly)、β-甘油磷酸鹽(β-gly)、單磷酸腺苷(amp)膽堿磷酸(pcho)和二酯磷(dna-p)),加d2o溶解并將溶液定容后配制成50mm(以磷計)的磷標準化合物溶液。
所需設備:
干冰或冰袋、冷凍干燥機、往復式振蕩機、離心機、電感耦合等離子體發射光譜儀(icp-oes)、安捷倫dd2500mhz或600mhz核磁共振儀(儀器配備5mmonenmr探頭)。
具體實施方式
實施例1
一種湖泊沉積物中單酯磷組分提取及其磷核磁共振譜圖分析方法,其包括如下步驟:
1、采集我國滇池表層沉積物(0-10cm)樣品,具體見表1,冷凍干燥后研磨粉碎過100目篩,裝入封口袋中,于0至-20℃低溫冷凍保存備用;
表1實施例1中湖泊表層沉積物總磷含量與金屬元素(al、ca、fe、mg和mn)含量特征
2、稱取1.00±0.01g沉積物樣品于50ml離心管中,加入8ml0.25m-50mmedta溶液,在室溫下(25℃)連續提取三次,每次提取時間分別依次為2h、4h和16h,將三次所得提取液于4℃,10000g條件下離心分離30min后收集、合并上清液,得提取液;
3、采用k2s2o8溶液消解后,分析測定提取液中總磷(tp)含量,或者直接采用電感耦合等離子體發射光譜儀(icp-oes)測定提取液中tp含量;
4、然后使用冷凍干燥機將提取液樣品冷凍干燥后得固體粉末,加入2mld2o使樣品充分溶解,得沉積物naoh-edta提取濃縮液樣品;
5、向步驟4中的提取濃縮液樣品中加入0.2ml1mna2s/d2o溶液,然后于4℃條件下,靜置沉淀17-20h后,10000g條件下,離心分離30min后得上清液即待測液1;
6、采用安捷倫dd2500mhz或600mhz核磁共振儀獲得待測液1的31p-nmr圖譜(圖1),其中,核磁共振儀的參數設置如下:溫度控制在25℃,脈沖為45℃(持續時間為5.3μs),采集時間為0.6s,馳豫時間設置為5s,掃描次數為12000~15000次;
7、配置濃度為50mm(以磷計)磷標準化合物溶液(直接溶解于d2o),具體磷標準化合物包括亞甲基二磷酸鹽(mdpa)、肌醇六磷酸鹽(myo-ihp)、葡萄糖6磷酸鹽(g6p)、葡萄糖1磷酸鹽(g1p)、α-甘油磷酸鹽(α-gly)、β-甘油磷酸鹽(β-gly)、單磷酸腺苷(amp)、膽堿磷酸(pcho)、以及二酯磷dna-p;將所配制的各磷標準化合物溶液分別移取20μl加入步驟1中的待測液1后測得31p-nmr譜圖b;
8、采用mestrenova(mnova)軟件(9.0.1版本,mestrelabresearchsl)處理31p-nmr譜圖,譜圖的展寬設置為2hz;調節31p-nmr譜圖a和b的正磷酸鹽位移至6.00ppm,初步比對譜圖a和b中單酯磷區域;然后精細對比譜圖a和b中單酯磷區域峰信號,確定沉積物中相同物質的峰2-3個;
9、基于譜圖b中標準的單酯有機磷化合物化學位移,調整譜圖a中的的單酯磷標準化合物化學位移,確定譜圖a中的單酯磷組分;
10、利用各單酯磷的峰面積比例及提取液中總磷的含量,計算各單酯磷的含量。測定結果如下表2。
表2實施例1湖泊沉積物naoh-edta提取及31p-nmr表征獲得單酯磷組分含量(mgkg-1)及其相對比例(%)
注:括號中值為各磷形態占naoh-edta提取液tp的百分比例。nuc:(mononucleotides)單核苷酸。
經測定,步驟5得到的待測1中各金屬離子去除率分別為:mn2+93%;fe3+98%;al3+96%;ca2+93%;mg2+91%。
對比例1
一種湖泊沉積物中單酯磷組分提取及其磷核磁共振譜圖分析方法,與實施例1相比,其區別僅在于:
步驟2、稱取1.00±0.01g沉積物樣品于50ml離心管中,一次性加入24ml0.25m-50mmedta溶液,在室溫下(25℃)提取22h,將提取液于4℃,10000g條件下離心分離30min后收集上清液,得提取液。
經測定,該實施例提取得到的各單酯磷的含量測定結果如下表3。
表3對比例1湖泊沉積物naoh-edta提取及31p-nmr表征獲得單酯磷組分含量(mgkg-1)及其相對比例(%)
注:“-”表示未檢測到該成分。
對比例2
一種湖泊沉積物中單酯磷組分提取及其磷核磁共振譜圖分析方法,與實施例1相比,其區別僅在于:
省略步驟7;
步驟8、采用mestrenova(mnova)軟件(9.0.1版本,mestrelabresearchsl)處理31p-nmr譜圖,譜圖的展寬設置為2hz;調節31p-nmr譜圖a的正磷酸鹽位移至6.00ppm;
步驟9、基于譜圖a中正磷酸鹽的化學位移6.00ppm,確定單酯磷化學位移,參考文獻報道《improvedpeakidentificationin31p-nmrspectraofenvironmentalsampleswithastandardizedmethodandpeaklibrary》(geoderma.257-258:102-114(2015))中公開的的31p-nmr譜圖中單酯磷的化學位移,以正磷酸鹽化學位移6.00ppm為相對位移,確定單酯磷組分。
經測定,該實施例提取得到的各單酯磷的含量測定結果如下表4。
表4對比例2與實施例1湖泊沉積物naoh-edta提取及31p-nmr表征單酯磷組分識別及其含量(mgkg-1)及其相對比例(%)比較結果
注:“-”表明未檢測到該成分。
由表4對比數據,結合上述實施例1中數據可以看出:單酯磷區域相同的峰,由于位移判斷方式的不同,導致不同的單酯磷組分、含量存在較大差異。實施例1分析得到的單酯磷與標準化合物的化學位移一致,結構及含量的測定結果可靠;而對比例2得到的結果與標準物質相差甚遠,導致對單酯磷化合物的結構及含量分析完全錯誤。例如,對比例2中分析得到的新單酯磷化合果糖6磷酸鹽(f6p)實際應為標準物質中的α-gly,同理,可以得出,對比例2中的g1p、nuc均為錯誤的分析結果,實際分別應為標準化合物中的scyllo-ihp、β-gly。
綜合來看,在樣品單酯磷化合物錯誤解析的情況下,分析結果完全錯誤。如對比例2中31p-nmr譜圖中單酯磷位移誤判,導致一些峰找不到對應的物質,如譜圖峰編號9對應的單酯磷化合物。再者,由于單酯磷化合物結構分析錯誤,單酯磷的含量也必然受到影響。如對比例2與實施例1獲得的含量上差異很大,如由于把β-gly判斷為nuc,其含量分析結果也由2.9mg/kg增加至9.1mg/kg,增加了近3倍。
上述例子僅作為說明的目的,本發明的范圍并不受此限制。對本領域的技術人員來說進行修改是顯而易見的,本發明僅受所附權利要求范圍的限制。
- 還沒有人留言評論。精彩留言會獲得點贊!