蝦夷扇貝中微囊藻毒素檢測方法與流程
本發明涉及一種蝦夷扇貝中微囊藻毒素檢測方法,屬于生化分離
技術領域:
,更具體涉及一種利用攪拌棒吸附分離蝦夷扇貝中7種微囊藻毒素并利用超高效液相色譜-質譜分析對其定性、定量分析的方法。
背景技術:
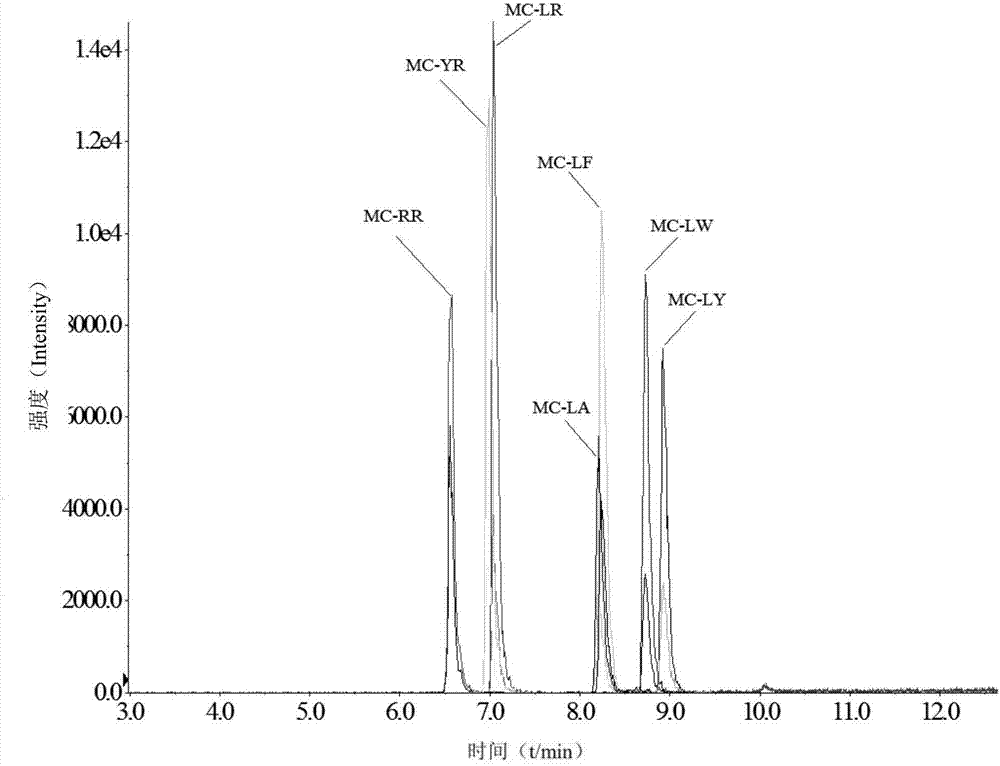
:微囊藻毒素(microcystins)為藍藻水華釋放的一類具有強烈致癌作用的肝毒素,其毒性較大,分布廣泛,目前已發現異構體多約80余種,其結構由7個氨基酸組成,主要產自微囊藻(microcystisaeruginosa)、魚腥藻(anabaena)、念珠藻(nostocales)等浮游性藍藻,其中mc-lr最為常見。調查表明,我國60%的水體由于過多接納了氮和磷等營養物質而出現了藍藻水華現象,該現象使水面湖靛堆積,大量消耗水中溶解氧致魚類窒息死亡,藻體死亡分解過程中釋放生物毒素類次級代謝產物,導致水體污染,水生動植物死亡。此外,已有研究證明,mcs進入人體肝細胞后,能強烈地抑制蛋白磷酸酶(pp1、pp2a)的活性,誘發細胞角蛋白高度磷酸化,導致肝細胞微絲分解、破裂和出血。因此,mcs通過食物鏈傳遞到水生動物中,并在水生動物體中積累富集而導致的人體健康潛在風險已經引起了國內外學者的普遍關注。因此建立一種淡水魚肉中高效、準確、靈敏的mcs分析檢測方法,對于水產品的質量安全與相關風險監測工作具有著重要意義。目前,對于各種生物體中mcs的檢測方法主要有酶聯免疫(elisa)法、聚合酶鏈式反應(pcr)、液相色譜(lc)法、液相色譜-串聯質譜(lc-ms/ms)法等,其中lc-ms/ms方法通量較大,定性能力強,更適用于復雜基質中mcs的殘留檢測。攪拌棒吸附萃取(sbse)是一種新型的固相微萃取樣品前處理技術,具有固定相體積大、萃取容量高、無需外加攪拌子、可避免競爭性吸附、能在自身攪拌的同時實現萃取富集等優點。目前廣泛采用的涂層為非極性的聚二甲基硅氧烷(pdms),這種涂層結構致密,且高度疏水、化學性質穩定,適用于水相中非極性和弱極性化合物的萃取。技術實現要素:本發明要解決的技術問題是提供一種靈敏度高的蝦夷扇貝中微囊藻毒素的檢測方法。為了解決上述技術問題,本發明提供了一種蝦夷扇貝中微囊藻毒素檢測方法,包括如下步驟:1)、蝦夷扇貝樣品前處理:①、取1g待測蝦夷扇貝肉進行勻漿;②、在漿液中加入10~20ml甲醇后于80±5℃超聲提取20~30min;③、將超聲提取所得物離心(冷凍離心機離心,即,控制溫度為3~5℃),取出上層清液;④、以離心所得的沉淀替代步驟②中的漿液重復上述步驟②~步驟③的超聲提取和離心;⑤、將所有的上層清液合并后用氮氣吹干,然后用5~10ml水復溶,得樣品液;2)、hlb/pdms涂層攪拌棒的制備:①、將攪拌棒清洗后烘干;②、配制pdms溶膠:將100±5μl聚二甲基硅氧烷(pdms)、100±5μl甲基三甲氧基硅烷(mtms)、10±1μl聚甲基氫硅氧烷(pmhs)和100±5μl95%三氟乙酸(tfa)混合,得pdms溶膠;95%三氟乙酸為由95%三氟乙酸和5%水混合而得,%為體積%;③將步驟①所得的烘干攪拌棒浸入步驟②所得的pdms溶膠(浸入時間約為5~10min),取出后在攪拌棒的表面裹上hlb微粒(親水親油平衡微粒,粒徑大小為10μl),然后于60~80℃干燥(烘箱烘干)24~30h,得hlb/pdms涂層攪拌棒;3)、攪拌棒吸附萃取(攪拌棒吸附萃取樣品中微囊藻毒素):先將hlb/pdms涂層攪拌棒在甲醇中浸泡20~30min(目的是去除表面的有機雜質,有機雜質指的是合成過程中使用的各種化合物),然后置入5~10ml步驟1)所得的樣品液中,加酸調整ph值為3~7(即,使吸附萃取過程中萃取環境的ph值為3~7),以600~1000rpm富集60~120min;然后取出hlb/pdms涂層攪拌棒放入含有100±20μl乙腈的離心管中超聲10~20min,使樣品中的mcs(微囊藻毒素)解吸附,所得到的解吸附溶液用于液相色譜-質譜分析;說明:上述用于調節ph值的酸是濃度為1~5mol/l的鹽酸溶液;4)、液相色譜-質譜條件(利用超高效液相色譜-質譜分析定性、定量分析樣品中微囊藻毒素):色譜柱:watersxselecthsst3(2.1mm×150mm,3.5μm),流動相:a為含0.1%甲酸的乙腈溶液,b為含0.1%甲酸的水溶液;梯度洗脫程序:0~1min,75%b;1~5min,75%~5%b;5~9min,5%b;9~10min,5%~75%b;10~16min,75%b;上述%均為體積%;柱溫40℃,流速0.30ml/min,進樣量10μl;離子源為電噴霧離子源,正源模式,離子源溫度500℃,電噴霧電壓5.5kv,霧化氣(gs1)壓力45psi,輔助氣(gs2)壓力65psi,氣簾氣(cur)壓力30psi,掃描方式為多反應監測(mrm)模式;7種微囊藻毒素的質譜參數見下表1;表1、7種微囊藻毒素的質譜參數注:rr等字母表示微囊藻毒素多肽中兩種可變氨基酸;從而獲得步驟3)所得的解吸附溶液中7種微囊藻毒素的濃度,最終經換算獲得蝦夷扇貝樣品7種微囊藻毒素的濃度。作為本發明的蝦夷扇貝中微囊藻毒素檢測方法的改進:所述步驟1)中,重復上述步驟②~步驟③的超聲提取和離心的次數為1~3次。作為本發明的蝦夷扇貝中微囊藻毒素檢測方法的進一步改進:所述步驟1)中,超聲為20khz,130w,50%變幅。作為本發明的蝦夷扇貝中微囊藻毒素檢測方法的進一步改進:所述步驟2)的①為將攪拌棒依次浸入水、二氯甲烷、1mol/lnaoh和0.1mol/lhcl中清洗(每次浸入的時間至少為8h),再用水沖洗,之后放入80~100℃烘箱烘干2~3h。作為本發明的蝦夷扇貝中微囊藻毒素檢測方法的進一步改進:所述步驟2)的③中,重復上述浸入pdms溶膠、取出后在攪拌棒的表面裹上hlb微粒,直至攪拌棒表面被hlb微粒完全包覆。作為本發明的蝦夷扇貝中微囊藻毒素檢測方法的進一步改進:步驟3)使用后的hlb/pdms涂層攪拌棒超聲清洗5~10min以便重復利用。在本發明步驟1)的樣品前處理中,利用電動攪拌器均質蝦夷扇貝樣品組織,用探頭式超聲(20khz,130w,50%變幅)提取。在本發明的步驟3)中,將富集后的攪拌棒取出后,用濾紙擦干表面再將其放入乙腈中超聲。本發明方法微囊藻毒素的提取效率高,有很好的特異性、準確度以及重復性,靈敏度較高,可以對蝦夷扇貝樣品中的微囊藻毒素進行良好的檢測,以確保公共衛生安全。綜上所述,本發明的方法利用攪拌棒吸附分離蝦夷扇貝中7種微囊藻毒素并利用超高效液相色譜-質譜分析對其定性、定量分析,能高效、精確地提取被測樣品中的微囊藻毒素,有良好的特異性、準確度以及重復性,檢測靈敏度較高。附圖說明下面結合附圖對本發明的具體實施方式作進一步詳細說明。圖1為hlb/pdms涂層攪拌棒表面形貌;(a)為放大倍數為18倍的掃描電子顯微鏡圖片;(b)為放大倍數為100倍的掃描電子顯微鏡圖片;(c)為放大倍數為500倍的掃描電子顯微鏡圖片;圖2為7種微囊藻毒素的總離子流色譜圖。具體實施方式下面結合具體實施例對本發明進行進一步描述,但本發明的保護范圍并不僅限于此。實施例1、一種蝦夷扇貝中微囊藻毒素檢測方法,依次進行以下步驟:1)、樣品前處理:利用電動攪拌器均質蝦夷扇貝樣品組織,取1g樣品(蝦夷扇貝肉)進行勻漿,加入10ml甲醇,加熱至80℃,并用探頭式超聲(20khz,130w,50%變幅)提取20min。之后用冷凍離心機離心(4℃、8000rpm離心15min)使固液分離,移出上層清液,并向沉淀中加入10ml甲醇重復上述的超聲提取、離心操作2次。將3次提取的上清液合并,用氮氣吹干,并用10ml水復溶;得樣品液。2)、攪拌棒制備:①將攪拌棒依次浸入水、二氯甲烷、1mol/lnaoh和0.1mol/lhcl中清洗(每次浸入的時間至少為8h),再用水沖洗,之后放入90℃烘箱烘干2h。所述攪拌棒是商品化的外涂層為聚二甲基硅氧烷(pdms)并內封一磁芯的玻璃管(10mm×0.5mm)。②將100μl聚二甲基硅氧烷(pdms)、100μl甲基三甲氧基硅烷(mtms)、10μl聚甲基氫硅氧烷(pmhs)和100μl95%三氟乙酸(tfa)混合,制得pdms溶膠。上述95%三氟乙酸(tfa)是指三氟乙酸(tfa)與水按照95:5的體積比進行混合。③將步驟①所得的攪拌棒浸入步驟②所得的pdms溶膠中7min,取出后在表面裹上hlb微粒(親水親脂平衡聚合物顆粒,粒徑大小為30μm),重復上述浸入、表面包裹步驟直至攪拌棒表面被hlb微粒完全包覆,將其放入65℃烘箱烘干24h。得hlb/pdms涂層攪拌棒。hlb微粒,例如可選用watersoasishlb微粒(30μm)。3)、攪拌棒吸附萃取使用前,將hlb/pdms涂層攪拌棒在甲醇中浸泡20min以去除表面的有機雜質。精確稱量5ml步驟1)所得的樣品液于eppendorf管中,將上述hlb/pdms涂層攪拌棒置于樣品液中,加酸(濃度為1mol/l的鹽酸溶液)調整ph值至5,以700~800rpm富集60~100min。將攪拌棒取出后,用濾紙擦干表面并將其放入含有100μl乙腈的離心管中超聲(探頭式超聲,20khz,130w,50%變幅)15min,使mcs解吸附,所得到的解吸附溶液可用于液相色譜-質譜分析。hlb/pdms涂層攪拌棒需超聲清洗(200w,60hz)5min以便重復利用。4)、液相色譜-質譜條件色譜柱:watersxselecthsst3(2.1mm×150mm,3.5μm),流動相:a為乙腈(含0.1%甲酸),b為水溶液(含0.1%甲酸),梯度洗脫程序為:0~1min75%b25%a1~5min75%~5%b25%~95%a5~9min5%b95%a9~10min5%~75%b95%~25%a10~16min75%b25%a柱溫40℃,流速0.30ml/min,進樣量10μl。離子源為電噴霧離子源,正源模式,離子源溫度500℃,電噴霧電壓5.5kv,霧化氣(gs1)壓力45psi,輔助氣(gs2)壓力65psi,氣簾氣(cur)壓力30psi,掃描方式為多反應監測(mrm)模式。7種微囊藻毒素的質譜參數見表2。表2、7種微囊藻毒素的質譜參數表3、7種微囊藻毒素的定量曲線方程和定量限說明:上述標準曲線中,x:質量濃度(μg/l);y:峰面積。y為實驗所得峰面積,根據上表中的標準曲線可以計算得到吸附溶液中該化合物的濃度x。因為解吸附是在100μl乙腈中,所以換算公式為x(μg/l)*100(μl)/0.5(g)即可得到蝦夷扇貝樣品中該化合物濃度。實驗一、步驟1)中,取7種微囊藻毒素原液以各0.1mg加入1ml乙腈中,混合均勻后得到微囊藻毒素標準品混合液,取該混合液10μl加入1g待測蝦夷扇貝肉勻漿;注:上述待測蝦夷扇貝肉是事先已確定的不含7種微囊藻毒素的樣品;步驟3)中,以800rpm富集100min。其余等同于實施例1。結果如表4:表4、7種微囊藻毒素的加標回收率及精密度(n=6)對比例1-1、將實驗一步驟1)中的提取20min改成重復提取10min;其余等同于實驗一。對比例1-2、將實驗一步驟1)中的重復提取2次改成重復提取0次;其余等同于實驗一。對比例2、將實驗一步驟2)中的將攪拌棒浸入pdms溶膠中7min改成浸入3min;其余等同于實驗一。對比例3-1、將實驗一步驟3)中的提取環境ph值5改成7;其余等同于實驗一。對比例3-2、將實驗一步驟3)中的富集提取100min改成富集提取30min;其余等同于實驗一。對比例4-1、將實驗一步驟4)中的特征子離子m/z135.0改為m/z134.0;其余等同于實驗一。對比例4-2、將實驗一步驟4)中的特征子離子m/z135.0改為m/z136.0;其余等同于實驗一。對比例4-3、將實驗一步驟4)中mc-rr的去簇電壓72ev改為40ev,mc-yr的去簇電壓66ev改為35ev,mc-lr的去簇電壓42ev改為25ev,mc-la、mc-lf、mc-lw的去簇電壓37ev改為20ev,mc-ly的去簇電壓32ev改為20ev;其余等同于實驗一。對比例4-4、將實驗一步驟4)中mc-rr的去簇電壓72ev改為90ev,mc-yr的去簇電壓66ev改為85ev,mc-lr的去簇電壓42ev改為60ev,mc-la、mc-lf、mc-lw的去簇電壓37ev改為55ev,mc-ly的去簇電壓32ev改為50ev;其余等同于實驗一。對比例4-5、將實驗一步驟4)中mc-rr的碰撞電壓50ev改為25ev,mc-yr、mc-lw、mc-ly的碰撞電壓25ev改為15ev,mc-lr、mc-la、mc-lf的碰撞電壓20ev改為10ev;其余等同于實驗一。對比例4-6、將實驗一步驟4)中mc-rr的碰撞電壓50ev改為60ev,mc-yr、mc-lw、mc-ly的碰撞電壓25ev改為30ev,mc-lr、mc-la、mc-lf的碰撞電壓20ev改為25ev;其余等同于實驗一。采用上述對比例所述方法所測得的各微囊藻毒素的回收率如表5所述。表5、各方法所述各微囊藻毒素回收率(%)綜上可見,本發明的蝦夷扇貝中微囊藻毒素檢測方法對于微囊藻毒素的提取效率高,有很好的特異性、準確度以及重復性,靈敏度較高,可以對蝦夷扇貝樣品中的微囊藻毒素進行良好的檢測,以確保公共衛生安全。此外,本發明提出的hlb/pdms涂層攪拌棒吸附萃取及超高效液相色譜-質譜聯用方法具有很大的潛力,為相關分析檢測提供了新的思路。實驗二、將如下表6所述的5種蝦夷扇貝按照實施例1所述方法進行檢測,所得結果如下表6所示。表6、本發明方法對每種蝦夷扇貝中各微囊藻毒素含量的檢測結果(μg/kg)驗證實驗、將實驗二中的5種蝦夷扇貝按照本行業所公認的精度最高的國標法進行檢查,所得結果如表7所示。表7、國標法對每種蝦夷扇貝中各微囊藻毒素含量的檢測結果(μg/kg)實驗三、關于特異性:步驟1)中,取實驗二中的蝦夷扇貝b組樣品,取7種微囊藻毒素原液與節球藻毒素原液以各0.1mg加入1ml乙腈中,混合均勻后得到8種毒素標準品混合液,取該混合液10μl加入上述樣品勻漿,步驟3)中,步驟3)中,以800rpm富集100min。其余等同于實施例1。所得結果見表8。表8、加入節球藻毒素后對于檢測微囊藻毒素含量的影響(μg/kg)最后,還需要注意的是,以上列舉的僅是本發明的若干個具體實施例。顯然,本發明不限于以上實施例,還可以有許多變形。本領域的普通技術人員能從本發明公開的內容直接導出或聯想到的所有變形,均應認為是本發明的保護范圍。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1