黃曲霉毒素B1適配體親和毛細管整體柱的制備及應用的制作方法
本發明涉及黃曲霉毒素b1的分離富集方法,具體涉及一種黃曲霉毒素b1適配體親和毛細管整體柱的制備方法及應用。
背景技術:
黃曲霉毒素b1(afb1)是主要由黃曲霉和寄生曲霉等產生的一種有毒代謝產物,已被國際癌癥研究機構(iarc)定為一級致癌物。黃曲霉毒素b1廣泛分布于各類農產品和食品中,其中最易受到污染的主要有花生、玉米、稻谷、小麥、花生油等糧油食品。鑒于afb1的嚴重危害性,國內外相關組織對其制定了嚴格的限量標準。
目前黃曲霉毒素b1的檢測方法主要有薄層色譜法(tlc)、高效液相色譜法(hplc)、液質聯用法(lc-ms)、免疫分析法(ia)等。在眾多的方法中,hplc由于操作簡單,準確可靠,重復性好,已成為實驗室內黃曲霉毒素b1的主要檢測方法。但是由于農產品和食品樣品基體復雜,在采用hplc檢測黃曲霉毒素b1時,需要對提取液有一個分離純化的過程。由于相對高的親和力和特異性,免疫親和柱在分離純化和檢測分析中得到了廣泛的應用。然而,在親和色譜中,免疫抗體存在一些明顯的局限性,(1)抗體在固定相上的固定化仍然具有一定的挑戰性,固定的抗體在空間上的取向難以保持一致,影響抗體的親和力;(2)抗體易受外界條件的影響,在強親和力的親和色譜模式下,分離富集操作過程中經常需要改變溶液的ph、添加有機溶劑或變性劑等,劇烈的洗脫條件,可能會導致抗體失活,在很大程度上限制了方法的靈活應用;(3)抗體由于體積大,在固定相表面的覆蓋率小,導致免疫親和柱容量比較小;(4)抗體制備需要經過免疫動物實驗或細胞實驗、繁瑣費時、成本高。
技術實現要素:
本發明的目的是針對上述問題,提供一種黃曲霉毒素b1適配體親和毛細管整體柱,將氨基修飾的黃曲霉毒素b1適配體通過戊二醛偶聯接枝修飾于有機-無機雜化的氨基毛細管整體柱,用于待測樣品中黃曲霉毒素b1的高選擇性分離與富集。
本發明的技術方案如下:一種黃曲霉毒素b1適配體親和毛細管整體柱的制備方法,包括如下步驟:
(1)毛細管活化:用于制備整體柱的石英毛細管內徑為530μm,外徑為690μm。分別用1.0mol/l氫氧化鈉溶液沖洗毛細管4h,去離子水沖洗30min,1.0mol/l鹽酸溶液沖洗4h,然后再以去離子水洗至中性,在160℃下用氮氣吹干。
(2)整體柱的原位合成:首先將10~30mgctab溶于150~350μl無水乙醇和50~150μl去離子水的混合溶液中。然后將100~200μlteos和50~100μlaptes快速加入上述混合溶液中。室溫渦旋0.5~5min,然后放置在-5~5℃的水浴中超聲0.5~5min后,將混合溶液灌入上述活化處理過的毛細管中。最后,毛細管兩端以硅橡膠封口,放置于30~60℃烘箱中反應10~30h。
(3)整體柱的清洗:反應完成后,以甲醇和水充分清洗毛細管。
(4)戊二醛衍生:將含有5~20%戊二醛的磷酸緩沖溶液以5~20μl/min速率通入毛細管整體柱中,連續反應12h,然后用磷酸緩沖溶液沖洗掉殘留的戊二醛。
(5)修飾適配體:將黃曲霉毒素b1適配體制成濃度為4~8nmol/l的磷酸緩沖溶液,注入毛細管整體柱中反應8~12h,重復三次,用磷酸緩沖溶液沖洗掉未反應的適配體。最后用3~8mg/ml氰基硼氫化鈉沖洗整體柱6h,將制得的毛細管整體柱截取5cm的長度用于之后的固相微萃取操作。
上述的毛細管整體柱在分離富集黃曲霉毒素b1中的應用。
一種基于上述的毛細管整體柱分離富集黃曲霉毒素b1的方法,它是基于黃曲霉毒素b1的毛細管整體柱選擇性捕捉上樣液中的黃曲霉毒素b1,實現黃曲霉毒素b1的分離富集;其步驟是:
(1)毛細管活化:用10~20mmol/l的tris-hcl緩沖溶液平衡親和毛細管整體柱,將親和毛細管整體柱活化;
(2)上樣:將待測樣品溶液以10~100μl/min通入親和毛細管整體柱內;
(3)清洗:待樣品全部流經親和毛細管整體柱后,用10~20mmol/ltris-hcl緩沖液將柱內殘留和未被特異性捕獲的黃曲霉毒素b1淋洗下來;
(4)洗脫:用含有40~60%甲醇的tris-hcl緩沖溶液將被親和毛細管整體柱捕獲的黃曲霉毒素b1洗脫。
本發明的黃曲霉毒素b1適配體親和毛細管整體柱,將氨基修飾的黃曲霉毒素b1適配體通過戊二醛偶聯接枝修飾于有機-無機雜化的氨基毛細管整體柱,通過適配體與黃曲霉毒素b1特異性親和作用,有機-無機雜化整體柱高的柱滲透性和大的比表面積,可以高靈敏、高選擇性地分離、富集復雜食品樣品中痕量、超痕量黃曲霉毒素b1,與檢測儀器聯用可實現黃曲霉毒素b1簡便、靈敏、快速檢測。
附圖說明
圖1本發明實施例1適配體親和毛細管整體柱的掃描電子顯微鏡圖;
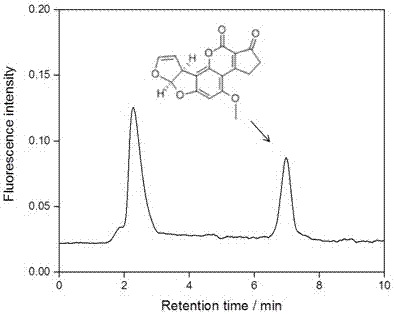
圖2本發明實施例4整體柱凈化后的色譜圖。
具體實施方式
本發明在有機-無機雜化毛細管整體柱的表面修飾并結合黃曲霉毒素b1適配體,然后用這種毛細管整體柱去分離富集黃曲霉毒素b1。以下實施例僅作為本發明內容的進一步說明,其中的實驗條件和設定參數不應視為本發明基本技術方案的局限。
實施例1黃曲霉毒素b1適配體親和毛細管整體柱的制備
(1)毛細管活化:用于制備整體柱的石英毛細管內徑為530μm,外徑為690μm。分別用1.0mol/l氫氧化鈉溶液沖洗毛細管4h,去離子水沖洗30min,1.0mol/l鹽酸溶液沖洗4h,然后再以去離子水洗至中性,在160℃下用氮氣吹干。
(2)整體柱的原位合成:首先將25mgctab溶于225μl無水乙醇和100μl去離子水的混合溶液中。然后將150μlteos和80μlaptes快速加入上述混合溶液中。室溫渦旋30s,然后放置在0℃的冰水浴中超聲30s后,將混合溶液灌入上述活化處理過的毛細管中。最后,毛細管兩端以硅橡膠封口,放置于40℃烘箱中反應20h。
(3)整體柱的清洗:反應完成后,以甲醇和去離子水充分清洗毛細管,以除去未反應的硅烷試劑和模板劑ctab。
(4)戊二醛衍生:配制含10%戊二醛的100mmol/l磷酸緩沖溶液(ph8.0),將該溶液以5μl/min的速率通入毛細管,連續反應12小時,然后用磷酸緩沖溶液沖洗掉殘留的戊二醛。
(5)適配體修飾:將黃曲霉毒素b1適配體溶于100mmol/l磷酸緩沖溶液,配制成5nmol/l的適配體溶液,然后以5μl/min速率通入毛細管,在4℃下反應8h,重復三次,用磷酸緩沖溶液沖洗掉未反應的適配體。最后用5mg/ml氰基硼氫化鈉溶液以5μl/min沖洗整體柱6h,將制得的整體柱截取5cm的長度用于之后的固相微萃取操作。
上述步驟中,黃曲霉毒素b1適配體的來源于實驗室合成,其堿基序列為:
5’-agcagcacagaggtcagatggtgctatcatgcgctcaatgggagactttagctg
cccccacctatgcgtgctaccgtgaa-3’,其中5'端通過c6間隔臂被-nh2修飾。
實施例2
首先采用與實施例1相同的步驟進行毛細管活化處理。然后,將10mgctab溶于150μl無水乙醇和50μl去離子水的混合溶液中,然后將100μlteos和50μlaptes快速加入上述混合溶液中。室溫渦旋5min,然后放置在-5℃的冰水浴中超聲5min后,將混合溶液灌入上述活化處理過的毛細管中。毛細管兩端以硅橡膠封口,放置于30℃烘箱中反應10h。反應完成后,以甲醇和去離子水充分清洗毛細管。
配制含5%戊二醛的磷酸緩沖溶液,將該溶液以10μl/min的速率通入毛細管,連續反應。在適配體修飾過程中,將黃曲霉毒素b1適配體制成濃度為4nmol/l的磷酸緩沖溶液,然后以5μl/min速率通入毛細管,在4℃下反應12h,重復三次,用磷酸緩沖溶液沖洗掉未反應的適配體。最后用3mg/ml氰基硼氫化鈉溶液以5μl/min沖洗整體柱6h,將制得的整體柱截取5cm的長度用于之后的固相微萃取操作。
實施例3
首先采用與實施例1相同的步驟進行毛細管活化處理。然后,將30mgctab溶于350μl無水乙醇和150μl去離子水的混合溶液中,然后將200μlteos和100μlaptes快速加入上述混合溶液中。室溫渦旋2min,然后放置在5℃的水浴中超聲2min后,將混合溶液灌入上述活化處理過的毛細管中。毛細管兩端以硅橡膠封口,放置于60℃烘箱中反應30h。反應完成后,以甲醇和去離子水充分清洗毛細管。
配制含20%戊二醛的磷酸緩沖溶液,將該溶液以20μl/min的速率通入毛細管,連續反應。在適配體修飾過程中,將黃曲霉毒素b1適配體制成濃度為8nmol/l的磷酸緩沖溶液,然后以5μl/min速率通入毛細管,在4℃下反應10h,重復三次,用磷酸緩沖溶液沖洗掉未反應的適配體。最后用8mg/ml氰基硼氫化鈉溶液以5μl/min沖洗整體柱6h,將制得的整體柱截取5cm的長度用于之后的固相微萃取操作。
實施例4黃曲霉毒素b1適配體親和毛細管整體柱的應用
(1)活化:用100μl10mmol/ltris-hcl平衡實施例1制備得到的親和整體柱,將親和整體柱活化。
(2)上樣:將500μl樣品溶液以10μl/min的速率通入整體柱內。
(3)清洗:待樣品全部流經親和柱后用50μl10mmol/ltris-hcl(ph=7.5)緩沖液將柱內殘留和未被特異性捕獲的黃曲霉毒素b1淋洗下來;
(4)洗脫:以20μl/min的速率,用含有40%甲醇的tris-hcl溶液,將被親和柱捕獲的黃曲霉毒素b1洗脫。
(5)上機檢測:采用高效液相色譜柱后衍生熒光檢測洗脫液中的黃曲霉毒素b1。色譜柱為c18反相色譜柱,檢測所采用的激發波長和發射波長分別為365nm和440nm。小麥加標樣品色譜分離圖如附圖2所示,其中橫坐標是保留時間,縱坐標是熒光強度。提取液加標量5μg/l濃度水平時,回收率為83.1~92.3%。
實施例5
采用與實施例4相同的步驟,對待測樣品中的黃曲霉毒素b1進行分離與富集,其不同點在于:其中采用20mmol/l的tris-hcl緩沖液,上樣速率為100μl/min,并用含60%甲醇的tris-hcl溶液進行洗脫處理。
本發明并不局限于上述實施方式,如果對本發明的改動或變型不脫離本發明的精神和范圍,倘若這些改動和變型屬于本發明的權利要求和等同技術范圍之內,則本發明也意圖包含這些改動和變型。
- 還沒有人留言評論。精彩留言會獲得點贊!