一種快速溶劑萃取?電霧式檢測器同時測定酸棗仁中四種成分的方法與流程
本發明涉及一種同時測定酸棗仁中多鐘成分的含量的方法,尤其是一種快速溶劑萃取-電霧式檢測器同時測定酸棗仁中四種成分的方法,屬于化學檢測技術領域。
背景技術:
酸棗仁為鼠李科棗屬植物酸棗ziziphusjujubamill.var.spinosa(bunge)huexh.f.chou的干燥成熟種子。具養心補肝,寧心安神,斂汗,生津之功效,常用于虛煩不眠,驚悸多夢,體虛多汗,津傷口渴。其主要的有效化學成分為三萜皂苷類、黃酮類、三萜類、生物堿等。具有鎮靜催眠、抗焦慮、抗驚厥、抗炎、抗腫瘤等作用。《中國藥典》2015年版一部酸棗仁含量測定的提取方法為傳統索氏提取法,該方法操作繁瑣、耗時長;快速溶劑萃取(ase)是指在密閉容器內于高溫(50~200℃)和高壓(500~3000psi)條件下,在短時間內用有機溶劑提取固體或半固體樣品的一種新型樣品前處理方法。與超聲、微波、回流、超臨界萃取等成熟方法相比,ase有萃取溶劑用量少、提取時間短、萃取效率高、操作簡單方便、安全和自動化程度高等優點。國外已有學者將這項技術應用于植物藥定量和定性分析中樣品的前處,但在中藥材有效成分的定量和定性分析中作為樣品的前處理方法應用較少,本研究嘗試將其應用到酸棗仁含量測定中以避免傳統方法的上述缺陷。
電霧式檢測器(cad)是一款新型的通用型檢測器,它基于霧化檢測器的原理,洗脫液經霧化后形成顆粒,經過漂移管干燥后與帶電氮氣碰撞,使得目標化合物帶上正電荷,最后通過靜電計測量電荷的量,該測量值與目標化合物的質量成一定比例關系,其響應不依賴于化合物的分子結構,能對大多數化合物提供一致響應性,同時能達到較高的靈敏度和低檢測極限、重現性、穩定性較好,因此能準確的用于定量分析。對于酸棗仁中有效成分的測定,傳統方法多采用液相二極管陣列檢測器、蒸發光檢測器和液相質譜檢測系統[15-16],然而酸棗仁皂苷a、b與白樺脂酸為三萜皂苷類成分,紫外吸收差,使用二極管陣列檢測器靈敏度低,而蒸發光散色檢測器(elsd)檢測基線不夠穩定、靈敏度也難以滿足要求;液相質譜檢測系統雖然靈敏度較高,但是儀器價格高、操作復雜、維護不易。電霧式檢測器是一種基于質量的靈敏檢測器,尤其適于測定與化學特性無關的任何不揮發分析物,該技術的靈敏度和精度比蒸發光散射檢測的高,重復性好,可達到納克級靈敏度,因此使用電霧式檢測器進行酸棗仁中多種有效成分的檢測具有很大的實際應用前景。
技術實現要素:
針對上述技術問題,本發明的目的是同時提供一種建立了同時測定酸棗仁中2種皂苷類、1種黃酮類和1種三萜類成分含量的方法,該方法高效準確、重現性好。
為解決上述技術問題,本發明的技術方案是這樣的:
一種快速溶劑萃取-電霧式檢測器同時測定酸棗仁中四種成分的方法,依次包括下述步驟:
1)對照品儲備液的制備
分別精密稱取酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸對照品適量,加甲醇溶解并定容至刻度,制成質量濃度分別為酸棗仁皂苷a4.1802mg.ml-1、酸棗仁皂苷b5.088mg.ml-1、斯皮諾素4.7304mg.ml-1、白樺脂酸4.272mg.ml-1的對照品儲備液,于冰箱中保存,備用;
2)供試品溶液的制備
取本品粉末1g,精密稱定,與1g硅藻土混合均勻填充于10ml萃取池,按ase正交設計優化的條件進行先除雜再提取,除雜過程:用正己烷在80℃靜態萃取8min,循環萃取1次,萃取液棄去并氮氣吹掃300s;提取過程:萃取溶劑為甲醇,萃取溫度100℃,靜態萃取時間8min,萃取循環次數為1次,沖洗體積60%,氮氣吹掃時間為60s;得到25ml萃取液回收溶劑至干,殘渣加甲醇溶解,轉移至5ml量瓶中,加甲醇至刻度,搖勻,濾過,取續濾液,即得;
3)色譜條件及系統適應性試驗
注入色譜儀檢測,色譜柱thermosyncronisc18(,流動相為乙腈-水,梯度洗脫程序為0~8min,10%~60%乙腈;8~10min,60%~80%乙腈;10~13min,80%~100%乙腈;13~16min,100%乙腈;流速0.5ml.min-1,柱溫40℃,霧化溫度35℃,進樣量1μl。
本發明與現有技術相比,具有以下優勢:
本發明提供的技術方案方法建立一種快速溶劑萃取(ase)-電霧式檢測器(cad)同時測定酸棗仁中酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸的方法。優化了ase參數,極大地提高了提取效率,通過1次進樣,同時測定酸棗仁中2種皂苷類、1種黃酮類和1種三萜類成分的含量,線性關系良好,相關系數為0.9983~0.9996,回收率在98.46%~102.02%之間。該方法快速、簡便、準確可靠,可用于酸棗仁藥材的質量控制。
附圖說明
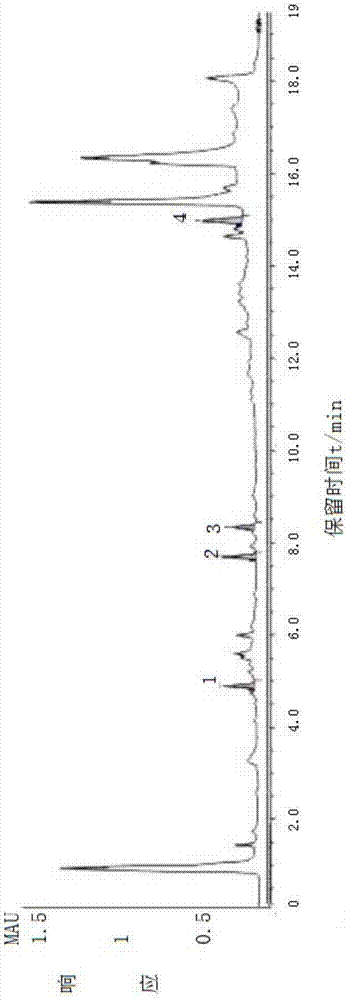
圖1混合對照品hplc圖
圖2是酸棗仁樣品的hplc圖。
具體實施方式
下面結合具體實施例對本發明的權利要求書做進一步說明,但不構成對本發明的任何限制,任何對本發明做有限次修改可得的技術方案仍然屬于本發明所要保護的范圍。
實施例1
1儀器與試藥
1.1儀器
thermofisherscitificultimate3000高效液相色譜儀配置電霧式檢測器coronaultra(賽默飛世爾科技公司)、ase350快速溶劑萃取儀(賽默飛世爾科技公司)、xa205du電子分析天平(梅特勒儀器有限公司),超純水機(默克密理博公司),ev311-v旋轉蒸發儀(北京萊伯泰科科技有限公司)。
1.2試藥
對照品酸棗仁皂苷a(批號110734-201512,含量以95.6%計)、酸棗仁皂苷b(批號110814-200506,含量以100%計)、斯皮諾素(批號111869-201203,含量以95.4%計)、白樺脂酸(批號111802-201001,含量以100%計)均由中國食品藥品檢定研究院提供;乙腈為色譜級,其余試劑均為分析純。
酸棗仁藥材均為市售品(批號1508015、1511018,產地河北;批號150401產地山東;批號150802、151110,產地江西;1511003、151008、151112,產地山東;1508003、151003,產地云南),經廣西梧州食品藥品檢驗所黃蘅副主任中藥師鑒定為鼠李科植物酸棗的干燥成熟種子。
2方法
2.1對照品儲備液的制備
分別精密稱取酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸對照品適量,加甲醇溶解并定容至刻度,制成質量濃度分別為酸棗仁皂苷a4.1802mg.ml-1、酸棗仁皂苷b5.088mg.ml-1、斯皮諾素4.7304mg.ml-1、白樺脂酸4.272mg.ml-1的對照品儲備液,于冰箱中保存,備用。
2.2供試品溶液的制備
取本品粉末(過四號篩)約1g,精密稱定,與約1g硅藻土混合均勻填充于10ml萃取池,按ase正交設計優化的條件進行先除雜再提取,除雜過程:用正己烷在80℃靜態萃取8min,循環萃取1次,萃取液棄去并氮氣吹掃300s;提取過程:萃取溶劑為甲醇,萃取溫度100℃,靜態萃取時間8min,萃取循環次數為1次,沖洗體積60%,氮氣吹掃時間為60s。得到約25ml萃取液回收溶劑至干,殘渣加甲醇溶解,轉移至5ml量瓶中,加甲醇至刻度,搖勻,濾過,取續濾液,即得。
2.3色譜條件及系統適應性試驗
色譜柱thermosyncronisc18(100mm×3mm,3μm,美國賽默飛世爾科技公司),流動相為乙腈-水,梯度洗脫程序為0~8min,10%~60%乙腈;8~10min,60%~80%乙腈;10~13min,80%~100%乙腈;13~16min,100%乙腈;流速0.5ml.min-1,柱溫40℃,霧化溫度35℃,進樣量1μl。各成分色譜峰計算的理論塔板數均≥4000。見圖1和圖2,其中:1-酸棗仁皂苷a,2-酸棗仁皂苷b,3-斯皮諾素,4-白樺脂酸。
3結果
3.1ase正交試驗及結果分析
3.1.1除雜溶劑和萃取溶劑的選擇
《中國藥典》2015年版采用石油醚作為除雜溶劑,本試驗同時考察了石油醚、正己烷的除雜效果,通過hplc-cad檢測對雜質峰高和數量進行比較,確定選擇正己烷為除雜溶劑,同時使用單因素考察確定除雜的ase條件:萃取溫度80℃,靜態萃取時間8min,萃取循環次數為1次,氮氣吹掃時間為300s以吹掃干正己烷。
根據待測成分的溶解性考察了甲醇、70%乙醇作為萃取溶劑,結果顯示甲醇與70%乙醇提取斯皮諾素含量一致,且雜質峰基本相同,考慮到甲醇更易于濃縮蒸干,故采用甲醇提取溶劑。
3.1.2ase萃取條件正交試驗的設計和結果分析
影響快速溶劑萃取的因素主要有萃取溫度、靜態萃取時間、沖洗體積和循環次數,本研究以酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸的萃取含量以及四種成分的萃取總量為考察指標,在單因素實驗中對萃取溫度(60℃~180℃)、沖洗體積(60%~100%)、靜態萃取時間(5min~10min)和循環次數(1~5次)進行初步篩選,確定沖洗體積為60%,進一步優化的萃取溫度范圍為80℃~120℃,靜態萃取時間為6min~10min,循環次數為1~3次。再采用l9(33)正交設計表優化,各試驗條件下重復2次。因素水平正交試驗設計表見表1,正交試驗結果分析見表2。表2的正交試驗結果可見,從各指標成分的極差來看,影響萃取效率最明顯的是溫度,其次是靜態萃取時間,影響各指標成分萃取含量較優水平依次為:斯皮諾素a2、b2、c1或c3;酸棗仁皂苷aa2、b1或b2、c1或c3;酸棗仁皂苷ba2、b1或b2、c1或c3;白樺脂酸a2、b3、c2;四種成分總量a2、b2或b3、c2。綜合指標成分的萃取效率及考慮節約溶劑和時間,最終選擇ase的萃取條件為萃取溶劑甲醇,萃取溫度100℃,靜態萃取時間8min,萃取循環次數為1次,沖洗體積60%,氮氣吹掃時間為60s。
表1l9(33)正交試驗設計ta
表2正交試驗結果分析
3.2線性關系考察
分別精密量取對照品儲備液0.4、0.8、1.2、1.6、2ml至10ml量瓶中,加甲醇至刻度,配制成一系列線性濃度,進樣量為1μl,在上述色譜條件下測定其峰面積,以各成分進樣量(μg)為橫坐標,峰面積(y)為縱坐標,繪制標準曲線,得回歸方程及相關系數,結果表明四種成分在0.2μg~1μg范圍內與峰面積呈良好線性關系,見表3。
表3酸棗仁中四種成分回歸方程和線性范圍
3.3精密度試驗
取上述對照品溶液(濃度約為0.4mg.ml-1),連續進樣6次,記錄峰面積,酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸峰面積的rsd依次為0.15%、0.41%、0.30%、0.23%、0.37%,表明儀器精密度良好。
3.4重復性試驗
取酸棗仁樣品(批號1508015),按“2.3”項下方法制備6份供試品溶液,按上述色譜條件進行測定,計算酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸含量分別為3.39mg.g-1、2.86mg.g-1、1.63mg.g-1、1.07mg.g-1,rsd依次為0.72%、1.02%、0.87%、0.51%,表明方法重復性良好。
3.5穩定性試驗
取重復性試驗供試品溶液,分別于0、4、8、12、16、20、24h進樣,記錄峰面積,計算酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸峰面積的rsd依次為0.38%、0.92%、0.43%、0.57%,表明供試品溶液在24h內穩定。
3.6加標回收率試驗
取已測定的酸棗仁樣品(批號1508015)6份,每份1g,精密稱定,精密加入酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸對照儲備液適量,按“2.3”項下方法制備供試品溶液,在上述色譜條件進行測定,分別計算酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸的平均回收率依次為102.02%、98.46%、101.16%、100.93%,rsd依次為1.82%、1.07%、1.93%、1.17%,表明該方法的準確度良好。
3.7檢出限和定量限的測定
取對照品溶液(濃度約為0.2mg.ml-1)逐級稀釋,測定酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸峰高,分別以信噪比(s/n)等于3和10為標準,測得4個待測成分的檢出限和定量限,本試驗方法檢出限酸棗仁皂苷a為0.7μg.ml-1、酸棗仁皂苷b0.8μg.ml-1、斯皮諾素0.8μg.ml-1、白樺脂酸0.7μg.ml-1;定量限酸棗仁皂苷a為2.4μg.ml-1、酸棗仁皂苷b2.6μg.ml-1、斯皮諾素2.6μg.ml-1、白樺脂酸2.4μg.ml-1。
3.8樣品測定以及不同提取方法的比較
取不同批次的酸棗仁粉末1g,精密稱定,分別按“2.2”項下以及《中國藥典》2015版一部酸棗仁項下方法制備供試品溶液,分別按各自方法色譜條件下進行分析測定,取兩種方法得到的含量結果進行t檢驗,根據t值表,所得t值<t(df)0.05=1.833,即兩種方法的統計學差異不顯著,結果見表4。
表410批樣品不同提取方法的含量比較
4討論
本研究采用快速溶劑萃取(ase)法進行酸棗仁中有效成分的提取,以酸棗仁皂苷a、酸棗仁皂苷b、斯皮諾素、白樺脂酸的萃取含量以及四種成分的萃取總量為指標,采用l9(33)正交試驗優化萃取溫度、靜態萃取時間和循環次數等ase的主要影響因素,與《中國藥典》傳統方法相比,除雜和提取過程能在一小時內完成,大大縮短了樣品前處理時間。兩種提取方法測得的各成分含量基本一致,快速溶劑萃取(ase)法操作方便,安全性好、自動化程度高并且環保。
cad為通用型檢測器,其檢測信號對不同結構的化合物有一致的響應,研究顯示,酸棗仁有效成分標準曲線在較小范圍內,cad檢測器的響應值與化合物質量呈直線關系,可采用外標兩點法計算,比較方便。與藥典方法中蒸發光檢測器(elsd)相比(檢測限約為20μg.ml-1),靈敏度更高,基線更穩定,利于定量分析。
目前對酸棗仁的質量研究只是局限于僅對酸棗仁皂苷類或斯皮諾素進行研究,本文建立的ase-hplc-cad方法能夠同時測定酸棗仁中2種皂苷類、1種黃酮類和1種三萜類成分的含量,拓展了酸棗仁質量研究的應用范圍。該方法簡便、快速、準確,可實現對酸棗仁多組分的質量控制,為ase延伸到中藥檢測的提取提供了理論基礎;同時也可利用cad特性,嘗試開發酸棗仁及其類似藥材的的一測多評方法用于全面評價和分析藥材的質量
以上所述的僅為本發明的較佳實施例,凡在本發明的精神和原則范圍內所作的任何修改、等同替換和改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!