一種Ag修飾的ZnO納米棒基底的自清潔傳感器及制備方法和用途與流程
本發明涉及一種ag修飾的zno納米棒基底的自清潔傳感器及制備方法和用途,屬功能材料制備技術領域。
背景技術:
目前,貴金屬納米粒子(mnps)材料,具有獨特的光學和電學性能,在傳感、催化和生物醫學等領域有廣泛的應用。
表面增強拉曼散射技術(sers),作為一種分析檢測技術,具有高靈敏度、快速響應和指紋譜圖等特性,已成為無損化學和生化分析最有價值的工具之一,近年來已經廣泛應用于化學/生物傳感器、單分子檢測、生物醫學檢測等領域。
在金屬納米粒子表面或接近表面的探針分子的拉曼信號能明顯放大108倍以上,使表面增強拉曼光譜技術可用于單分子的檢測與分析。更重要的是,銀納米粒子比金納米粒子具有更強和更靈敏的拉曼信號,因此成為更具有競爭力的等離子體傳感器并且拓寬了增強表面增強拉曼散射(sers)的應用范圍。然而,使用金屬納米粒子的主要缺點是:重復性和穩定性相對較低,特別是為提高靈敏度添加聚合劑的時候,重復性和穩定性會更低。重要的是,傳統的sers基底不能輕易清洗以及再生后重復用于表面增強拉曼光譜分析。
最近,一些報道都集中于金屬納米粒子在固體基質上活性的研究,如越來越多的玻璃組件、硅晶片和金屬半導體甚至是用濾紙調整的基底。一方面,它能解決傳統金屬納米基底含有的缺陷,進一步促進和拓寬sers應用。另一方面,一些調查報道,發現金屬納米顆粒與金屬半導體接觸時拉曼信號顯著增強。例如,有人制備三維al2o3膜負載金納米粒子探索sers增強機理。還有人制備表面用agnps修飾的活性相tio2納米纖維用于4-mba的檢測。認為消逝場的增加可以增強的sers效應,增強金屬納米粒子或金屬/半導體之間的關聯效應,可使特定的金屬半導體界面的相互作用增強。此外,最近的研究證明,金屬半導體也能產生弱sers信號。
最近,分子印跡技術(mit)與sers檢測技術聯用,提高了傳統的sers基底材料的選擇性。關于分子印跡聚合物(mips)基于表面增強拉曼光譜的研究已經提出,例如:胡亞錫等人制備的新型生物傳感器結合分子印跡聚合物和表面增強拉曼光譜測定牛奶中的三聚氰胺。kamra等人建立了基于表面增強拉曼光譜分子印跡聚合物,用于檢測尼古丁。雖然這些方法對sers的檢測具有諸多優點,但是關于貴金屬與半導體相結合,作為異質結基底材料并結合mips用于sers檢測的研究仍鮮有報道。
在本發明中,選取異質結構的sers基底結合mips用于檢測水中羅丹明6g,從而提高了選擇性。一般來說,羅丹明6g(r6g)作為模板分子,首先嘗試制備基于氧化鋅/銀異質結構作為sers基底的mips。在最佳條件下,r6g為模板分子,氧化鋅/銀作為sers基底,丙烯酰胺(am)為功能單體、乙二醇二甲基丙烯酸酯(egdma)為交聯劑,通過沉淀聚合制備具有高靈敏度的氧化鋅/銀/r6g的分子印跡聚合物。制備的此基底可以選擇性吸附r6g,顯著提高了r6g的拉曼信號。此外研究表明,zoa-mips在紫外光照射下具有自潔的性能。傳感裝置可以實現對r6g快速檢測,這種技術提供一個有吸引力的和具有成本效益的方法,拓寬了sers檢測的應用范圍。
技術實現要素:
本發明的目的是通過三步反應合成ag修飾的zno納米棒基底的自清潔傳感器,并探索其應用。首先,將氯化鋅(zncl2)分散到去離子水中,在室溫下超聲,在攪拌條件下滴加nh3·h2o。將混合液轉移到高壓釜中反應數小時。反應后,反復洗滌,離心分離,烘干備用;其次,將氧化鋅納米棒(znonrs)分散在乙醇和水的混合溶液中,加入0.1moll-1的agno3溶液和0.2moll-1的pvp,避光條件下磁力攪拌數小時后,加入ea,反應數小時,真空干燥待用;最后,將zno/ag分散到甲苯和mps的混合溶液中,然后通n2。數小時后,將合成產物離心分離;將zno/ag分散到乙腈中,充分超聲,然后將r6g、am和egdma加入溶液中,通n2數分鐘。最后,將aibn加入到混合溶液中,放入恒溫水浴中振蕩,采用兩步聚合方法制備。
本發明采用的技術方案如下:
一種ag修飾的zno納米棒基底的自清潔傳感器,所述傳感器是由zno納米棒、ag、印跡層復合而成的,ag負載在zno納米棒表面,形成zno/ag;印跡層是由丙烯酰胺(am)、乙二醇二甲基丙烯酸酯(egdma)和偶氮二異丁腈(aibn)聚合而成的,所述印跡層負載在zno/ag表面,形成ag修飾的zno納米棒基底的自清潔傳感器;所述ag修飾的zno納米棒基底的自清潔傳感器對羅丹明6g具有選擇性吸附的特性。
一種ag修飾的zno納米棒基底的自清潔傳感器的制備方法,步驟如下:
步驟1、zno納米棒的制備
將zncl2分散到去離子水中,在室溫下超聲,在攪拌條件下滴加nh3·h2o,將得到的混合液轉移到高壓釜中,在140~160℃進行水熱反應;反應完畢后,將合成的產物離心分離,洗滌,真空干燥,得到zno納米棒,記為znonrs;待用;
步驟2、zno/ag的制備
將zno納米棒分散在乙醇/水混合溶液中,加入0.1moll-1的agno3溶液和0.2moll-1的pvp溶液,避光條件下磁力攪拌,加入ea(丙烯酸乙酯),溫度升高到40~60℃反應11-13h;反應完畢后,將合成的產物離心分離,洗滌,真空干燥,得到zno/ag;待用;
步驟3、ag修飾的zno納米棒基底的自清潔傳感器的制備
將zno/ag分散到甲苯/mps混合液中80~100℃下通足量惰性氣體以排除溶液中氧氣;11-13h后,將固體離心分離,洗滌,真空干燥,得到固體a,待用;
將固體a分散到乙腈中,充分超聲,然后將r6g、am和egdma加入到溶液中,通惰性氣體以排除溶液中氧氣;最后,將aibn加入到混合溶液中,放入恒溫水浴中振蕩,得到ag修飾的zno納米棒基底的自清潔傳感器,產物記為zoa-mips。
步驟1中,所述zncl2與去離子水的用量比為1g:60~80ml;nh3·h2o與去離子水的體積比為5.0ml:60-80ml;所述水熱反應的時間問2.0-4.0h。
步驟2中,zno納米棒、乙醇/水混合溶液、agno3溶液和pvp溶液的用量比為100mg:40~60ml:1~3ml:4~6ml,所述agno3溶液的濃度為0.1moll-1,所述pvp溶液的濃度為0.2moll-1;所述zno納米棒與所述ea的用量比為100mg:200~400μl;所述磁力攪拌的時間為3.0-5.0h;所述溫度升高到40~60℃反應的時間為11-13h。
步驟3中,制備固體a時,zno/ag與甲苯/mps混合液中的mps用量比為1.0g:1.0~3.0ml,甲苯/mps混合液中,甲苯與mps的體積比為40~60:1~3;制備ag修飾的zno納米棒基底的自清潔傳感器時,固體a、乙腈、r6g、am、egdma、aibn的用量比為1.0g:50~70ml:0.1~0.3mmol:0.3~0.5mmol:1.0~3.0mmol:9.0~11mg。
步驟1~3中,所述的洗滌,均為乙醇和水分別洗滌3次。
所述的ag修飾的zno納米棒基底的自清潔傳感器用于選擇性吸附羅丹明6g。
本發明對應的非印跡聚合物的制備方法類似合成方法如上,但不加r6g,產物記為zoa-nips。
本發明的技術優點:
本發明將拉曼檢測技術與分子印跡技術相結合,使其產物具有靈敏的檢測性與高度的選擇性;在本發明中,將用生物低毒性的銀納米粒子修飾的氧化鋅基底與分子印跡技術相結合,使其聚合產物具有良好的生物兼容性,能夠應用于對羅丹明的檢測。銀納米粒子比金納米粒子具有更強和更靈敏的表面增強拉曼信號,使它們成為更具有競爭力的等離子體傳感器拓寬了表面增強拉曼散射(sers)的應用。
附圖說明
圖1:zno納米粒子(a),zno-ag(b)和zoa-mips(c)(比例尺尺寸為1μm)的掃描電鏡圖;
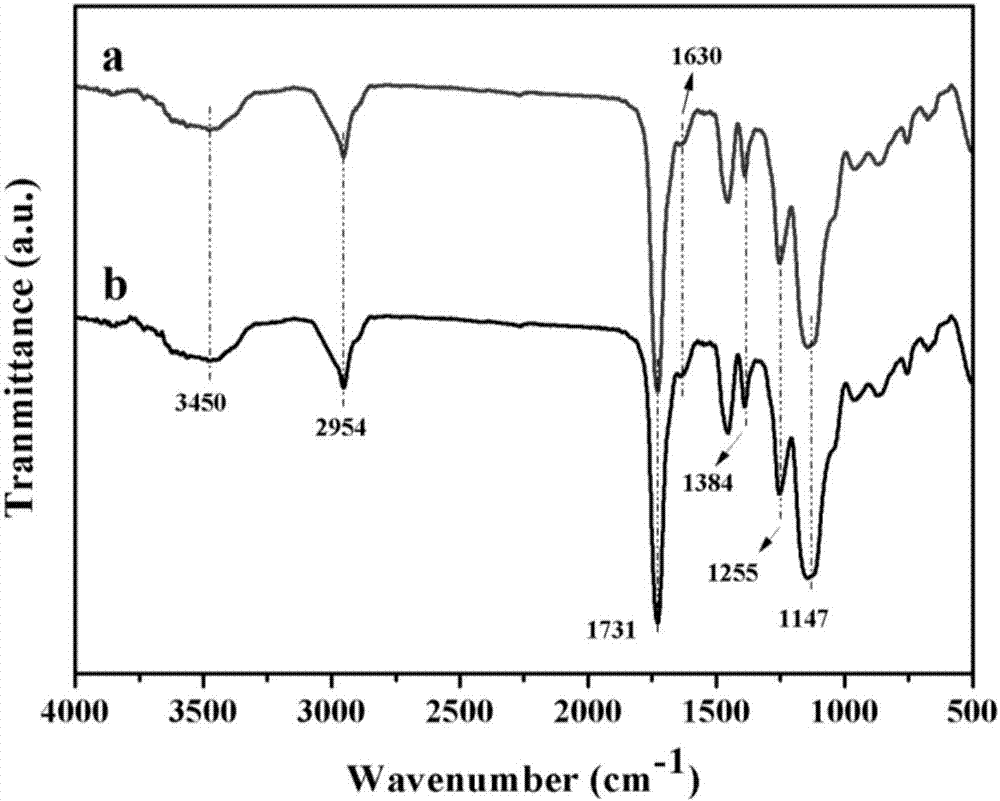
圖2:zoa-mips(a)和zoa-nips(b)的紅外光譜圖;
圖3:zoa-mips和zoa-nips的吸附性能顯示圖;
圖4:zoa-mips對于不同濃度r6g檢測的拉曼光譜(a)和拉曼強度與r6g濃度變化的檢測線性關系圖(b);
圖5:zoa-mips的選擇性檢測性能力圖(a:r6g檢測曲線;b:rb檢測曲線;c:cv檢測曲線);
圖6:zoa-mips的重復檢測性能。
具體實施方式
下面結合具體實施實例對本發明做進一步說明。
實施例1:
(1)zno納米棒的合成
在150ml單口燒瓶中,將1.0g氯化鋅分散到60ml的去離子水中,在室溫下超聲,在攪拌條件下滴加5.0mlnh3·h2o。將混合液轉移到高壓釜中140℃反應2.0h。隨后,將合成的產物離心分離,反復洗滌數次,真空干燥待用。
(2)zno/ag的合成
在150ml單口燒瓶中,將100mgznonrs分散在40ml乙醇和水(體積比4/1)的混合溶液中,加入1.0ml濃度為0.1moll-1的agno3溶液和4.0ml濃度為0.2moll-1的pvp,避光條件下磁力攪拌3.0h后,加入200μlea,溫度升高到40℃反應11h。隨后,將合成的產物離心分離,用乙醇和水反復洗滌三次,真空干燥待用。
(3)zno/ag分子印跡聚合物的制備
在150ml單口燒瓶中,將1.0gzno/ag分散到40ml甲苯和1.0mlmps的混合溶液80℃下通足量n2以排除溶液中氧氣。11h后,將合成產物離心分離,乙醇洗滌三次,真空干燥待用。
在150ml單口燒瓶中,將1.0gzno/ag分散到50ml乙腈中,充分超聲,然后將0.1mmolr6g、0.3mmolam和1.0mmolegdma加入溶液中,通足量n2以排除溶液中氧氣。最后,將9mgaibn加入到混合溶液中,放入恒溫水浴振蕩器,采用兩步聚合方法制備。
其中,步驟(1)所述的反應體系中,nh3·h2o與去離子水的體積比為5.0ml:60ml。步驟中所述的洗滌,均為乙醇和水分別洗滌3次。
步驟(2)所述的反應體系中,zno與agno3質量體積比為100mg:1.0ml,zno與混合溶液的質量體積比為100mg:40ml,zno與pvp的質量體積比為100mg:4.0ml,zno與ea的質量體積比為100mg:200μl。
步驟(3)所述的反應體系中,zno/ag與mps的質量體積比為1.0g:1.0ml,zno/ag與甲苯的質量體積比為1.0g:40ml,zno/ag與乙腈的質量體積比為1.0g:50ml。步驟中所述的洗滌,均為乙醇和水分別洗滌3次。
本發明對應的非印跡聚合物的制備方法類似合成方法如上,但不加r6g。
實施例2:
(1)zno納米棒的合成
在150ml單口燒瓶中,將1.0g氯化鋅分散到70ml的去離子水中,在室溫下超聲,在攪拌條件下滴加5.0mlnh3·h2o。將混合液轉移到高壓釜中150℃反應3小時。隨后,將合成的產物離心分離,反復洗滌數次,真空干燥待用。
(2)zno/ag的合成
在150ml單口燒瓶中,將100mgznonrs分散在50ml乙醇和水(體積比4/1)的混合溶液中,加入2.0ml濃度為0.1moll-1的agno3溶液和5.0ml濃度為0.2moll-1的pvp,避光條件下磁力攪拌4h后,加入300μlea,溫度升高到50℃反應12h。隨后,將合成的產物離心分離,用乙醇和水反復洗滌三次,真空干燥待用。
(3)zno/ag分子印跡聚合物的制備
在150ml單口燒瓶中,將1.0gzno/ag分散到50ml甲苯和2.0mlmps的混合溶液90℃下通足量n2以排除溶液中氧氣。12h后,將合成產物離心分離,乙醇洗滌三次,真空干燥待用。
在150ml單口燒瓶中,將1.0gzno/ag分散到60ml乙腈中,充分超聲,然后將0.2mmolr6g、0.4mmolam和2.0mmolegdma加入到溶液中,通足量n2以排除溶液中氧氣。最后,將10mgaibn加入到混合溶液中,放入恒溫水浴中振蕩,采用兩步聚合方法制備。
其中,步驟(1)所述的反應體系中,nh3·h2o與去離子水的體積比為5.0ml:70ml。步驟中所述的洗滌,均為乙醇和水分別洗滌3次。
步驟(2)所述的反應體系中,zno與agno3質量體積比為100mg:2.0ml,zno與混合溶液的質量體積比為100mg:50ml,zno與pvp的質量體積比為100mg:5.0ml,zno與ea的質量體積比為100mg:300μl。
步驟(3)所述的反應體系中,zno/ag與mps的質量體積比為1.0g:2.0ml,zno/ag與甲苯的質量體積比為1.0g:50ml,zno/ag與乙腈的質量體積比為1.0g:60ml。步驟中所述的洗滌,均為乙醇和水分別洗滌3次。
本發明對應的非印跡聚合物的制備方法類似合成方法如上,但不加r6g。
實施例3:
(1)zno納米棒的合成
在150ml單口燒瓶中,將1.0g氯化鋅分散到80ml的去離子水中,在室溫下超聲,在攪拌條件下滴加5.0mlnh3·h2o。將混合液轉移到高壓釜中160℃反應4.0h。隨后,將合成的產物離心分離,反復洗滌數次,真空干燥待用。
(2)zno/ag的合成
在150ml單口燒瓶中,將100mgznonrs分散在60ml乙醇和水(體積比4/1)的混合溶液中,加入3.0ml濃度為0.1mol/l的agno3溶液和5.0ml濃度為0.2moll-1的pvp,避光條件下磁力攪拌5.0h后,加入400μlea,溫度升高到60℃反應13h。隨后,將合成的產物離心分離,用乙醇和水反復洗滌三次,真空干燥待用。
(3)zno/ag分子印跡聚合物的制備
在150ml單口燒瓶中,將1.0gzno/ag分散到60ml甲苯和3.0mlmps的混合溶液100℃下通足量n2以排除溶液中氧氣。13h后,將合成產物離心分離,乙醇洗滌三次,真空干燥待用。
在150ml單口燒瓶中,將1.0gzno/ag分散到60ml乙腈中,充分超聲,然后將0.3mmolr6g、0.4mmolam和3.0mmolegdma加入溶液中,通足量n2以排除溶液中氧氣。最后,將11mgaibn加入到混合溶液中,放入恒溫水浴中振蕩,采用兩步聚合方法制備。
其中,步驟(1)中所述的反應體系中,nh3·h2o與去離子水的體積比為5.0ml:80ml。步驟中所述的洗滌,均為乙醇和水分別洗滌3次。
步驟(2)所述的反應體系中,zno與agno3質量體積比為100mg:3.0ml,zno與混合溶液的質量體積比為100mg:60ml,zno與pvp的質量體積比為100mg:6.0ml,zno與ea的質量體積比為100mg:400μl。
步驟(3)所述的反應體系中,zno/ag與mps的質量體積比為1.0g:3.0ml,zno/ag與甲苯的質量體積比為1.0g:60ml,zno/ag與乙腈的質量體積比為1.0g:70ml。步驟中所述的洗滌,均為乙醇和水分別洗滌3次。
本發明對應的非印跡聚合物的制備方法類似合成方法如上,但不加r6g。
圖1:zno納米粒子(a),zno-ag(b)和zoa-mips(c)的掃描電鏡圖。所述傳感器是由zno納米棒、ag、印跡層復合而成的,ag負載在zno納米棒表面,形成zno/ag;印跡層是由丙烯酰胺(am)、乙二醇二甲基丙烯酸酯(egdma)和偶氮二異丁腈(aibn)聚合而成的,所述印跡層負載在zno/ag表面,形成ag修飾的zno納米棒基底的自清潔傳感器,該印跡聚合物已經成功合成;
圖2:zoa-mips(a)和zoa-nips(b)的紅外光譜圖。由圖中可以看出,這兩種材料都呈現出明顯的峰值,從紅外譜圖中,可以看出,該聚合物已經成功合成;
圖3:zoa-mips和zoa-nips的吸附性能顯示圖。在紫外光譜圖中,zoa-mips和zoa-nips吸附效率分別為71.23%和30.26%;
圖4:zoa-mips對于不同濃度r6g檢測的拉曼光譜(a)和拉曼強度與r6g濃度變化的檢測線性關系圖(b)。
圖5:zoa-mips的選擇性檢測性能力圖。r6g有明顯的拉曼增強信號,但對于rb和cv的拉曼強度比r6g小;
圖6:zoa-mips的重復檢測性能,從圖中可以看出,zoa-mips具備良好的可重復性能。
本發明具體的拉曼檢測按照下述方法進行:在拉曼光譜中反應出sers對zoa-mips的檢測能力。在本實驗中,所有的拉曼檢測條件均一致:激發光波長為633nm。每個樣品的光譜收集與曝光時間均為10秒,入射激光的功率為0.25mw。sers譜圖用50×尼康鏡頭收集。所有的sers基底放在載玻片上,自然干燥后用于表面增強拉曼光譜的檢測。
試驗例1:如圖6(a)所示,檢測了zoa-mips的sers活性,羅丹明6g為探針分子,確定檢測限。該圖表明,在1654cm-1處的表面增強拉曼光譜強度最強。此外,根據文獻可知,由于有對稱形式的c-c伸縮振動存在,所以將1654cm-1作為特征峰。從數據中可以看出,隨著r6g濃度從10-7moll-1到10-13moll-1的變化,sers的強度也隨之變化。當r6g濃度為10-14moll-1,拉曼信號幾乎消失。此外,如圖6(b)所示,特征峰強度的變化與r6g濃度的改變呈線性關系。r6g濃度在10-7moll-1到10-13moll-1之間時(r2)的檢測系數為0.95。
試驗例2:為了研究zoa-mips對r6g的特異選擇性,用濃度為10-7moll-1結構類似于羅丹明6g的rb和cv做平行實驗來進一步研究。如圖2所示,由于rb和cv分子結構與r6g不同,不能被zoa-mips選擇性吸附,所以只能觀察到微弱的表面增強拉曼光譜強度。
試驗例3:傳統的sers傳感器無法實現循環利用,所以sers傳感器的檢測效果受到限制。因此需要一個方法來制造一個用于檢測r6g的可再生使用的sers傳感器。在該試驗中制備的材料不僅擁有檢測功能,而且擁有自清潔功能。圖4中所示,在紫外光照射下,制備的sers基底對r6g的光催化活性的降解進行了研究。研究表明,該材料具有良好的光催化活性。在圖5中所示,通過適當的漂洗和光降解過程,zoa-mips中檢測不到有模板分子殘留。這證明了模板分子從mips的表面完全除去了。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種磁性納米粒子與超濾膜結合處理廢棄乳化液的方法與流程
- 一種多肽修飾納米粒子在制備治療急性肺損傷的藥物中的應用的制作方法與工藝
- 一種金納米花修飾的離子液體功能化石墨烯紙電極、其制備方法和應用與流程
- 一種新型的透明質酸修飾的多級納米顆粒及其制備與應用的制作方法與工藝
- 一種核殼結構的磁性納米粒子及其制備與應用的制作方法與工藝
- 一種室溫固相反應制備的納米鐵修飾的鐵基氨合成催化劑及其制備方法和應用與流程
- 一種丁氧基修飾的TiO2單晶空心四方納米錐材料、制備方法及其應用與流程
- 一種改性粒子修飾的有機無機雜化聚合物涂料的制作方法與工藝
- 一種表面修飾片狀納米銅及含有該納米銅的復合潤滑油的制備方法與流程
- 一種用于簡便修飾磁性納米粒子的手性拆分添加劑的制備和應用的制作方法與工藝
- 使用經修飾的核苷酸和納米孔檢測的DNA邊合成邊測序的制作方法與工藝
- 一種離子液體修飾的硫化鋅納米極壓抗磨劑的制備及含有該抗磨劑的節能抗磨液壓油的制作方法與工藝
- 一種Ag修飾的ZnO納米棒基底的自清潔傳感器及制備方法和用途與流程
- 一種Pt納米粒修飾ZnSnO3納米片氣敏材料的制備方法與流程
- 一種載有阿霉素的適配體修飾DNA納米籠及其制備方法與流程
- 使用經修飾的核苷酸和納米孔檢測的DNA邊合成邊測序的制造方法與工藝
- 一種鞋面填充上光修飾劑及其制備方法與流程
- 一種以碳納米點界面修飾的鈣鈦礦太陽能電池及其制備方法與流程
- 一種通過熒光分子修飾的銀溶膠檢測Cd2+、Pb2+離子的方法與流程
- 釓修飾硫化鉍納米診療劑的制備方法與制造工藝
- 一種表面修飾片狀納米銅及含有該納米銅的復合潤滑油的制備方法與流程
- 多肽修飾的納米粒子及其制備方法和控制細胞中基因表達的方法與流程
- 一種表面修飾片狀納米銅及含有該表面修飾片狀納米銅的潤滑油的制作方法與工藝
- 一種山核桃仁鞣質葉酸修飾納米粒的制備方法與流程
- 一種SiO2修飾TiO2單晶粒子光催化劑的合成方法與流程
- 一種用β-PbO2粒子修飾碳纖維的方法及其應用與流程
- 一種Ag修飾的ZnO納米棒基底的自清潔傳感器及制備方法和用途與流程
- 抗生素修飾的磁性納米粒子快速富集分離單增李斯特菌的方法與流程
- 一種水溶性透明質酸修飾的金納米粒子聚集體HA?Cys?AuNPs及制備方法與流程
- 一種多維度納米粒子修飾的超疏水吸油泡沫材料及其制備方法與流程