用于離子色譜中的極弱酸檢測的滲透性胺或酸引入的制作方法
本發明涉及目的弱解離分析物的離子色譜和增強檢測。
背景技術:
抑制電導陰離子色譜(scac)應用范圍從半導體制造中的痕量分析i到藥物分析ii,僅舉幾例。雖然scac擅長于測量強酸性陰離子,但弱酸性陰離子響應不良或完全不響應。盡管具有pka<7的酸顯示一定響應,但它們以非線性方式顯示響應。來自極弱酸(pka≥7.0)的陰離子例如硅酸鹽或氰化物基本上無法測量。
本發明涉及使用離子色譜("ic")的方法和儀器,其中抑制的分析物在檢測前轉換為鹽。
離子色譜是用于分析離子的已知技術,其通常包括使用含有電解質的洗脫劑的色譜分離區和洗脫劑抑制階段,隨后為通常通過電導檢測器執行的檢測。在色譜分離階段,注入樣品的離子從分離柱中洗脫。在抑制階段,洗脫劑電解質的電導率而不是分離的離子的電導率被抑制,如果分離的離子來源于強酸或堿。在第一代離子色譜中,電解質的抑制或剝離使用離子交換樹脂床。在改善形式的抑制中,使用以纖維或片材形式的荷電膜代替樹脂床。在片材形式中,樣品和洗脫劑在片材的一側上通過,而在片材的另一側具有流動的再生劑。該片材包括分配來自色譜分離的流出物的再生劑的離子交換膜。膜通過與膜的可交換離子相同電荷的離子,以將洗脫劑的電解質轉換成弱離子化形式,隨后為離子的檢測。
一種有效形式的抑制器在美國專利號4,999,098中描述。在這種儀器中,抑制器包括由離子交換膜片隔開的至少一個再生劑區室和一個色譜流出物區室。該片材允許與其可交換離子相同電荷的離子的跨膜通過。離子交換篩用于再生劑和流出物區室。來自流出物區室的流動被導向檢測器,例如電導率檢測器,用于檢測分辨的離子種類。篩提供離子交換部位且作用于提供跨越流出物流動通道的位點至位點轉移路徑,使得抑制能力不再受離子從總體溶液擴散到膜的限制。還公開了夾心抑制器,其包括與第一膜片相對且限定第二再生劑區室的第二膜片。公開了沿抑制器的長度與兩個再生劑室連通的間隔開的電極。通過跨越電極應用電勢,存在裝置的抑制能力中的增加。該專利公開了在再生劑流動通道中流動且由再生劑遞送源供應的通常再生劑溶液(酸或堿)。在通常的陰離子分析系統中,氫氧化鈉是洗脫劑且硫酸是再生劑。該專利還公開了使用水來替換電滲析模式中的再生劑溶液。在美國專利號5,352,360中所述的改善形式的膜抑制器中,來自檢測器的流出物再循環通過再生劑流動通道。
在berglundi.等人anal.chem.63:2175(1991)中,描述了另一個多重檢測器系統。此處,使用第一電導檢測器執行常規ic。來自該檢測器的流出物序貫經過陽離子交換和陰離子交換轉換區。對于陰離子分析,當從抑制器離開時,來自第一檢測器的流出物是通常ic形式的hx(其中x是分析物陰離子)。公開了兩個不同類型的轉換器。在序貫填充柱形式中,流出物首先通過陽離子(鈉)交換樹脂,并且隨后通過陰離子(氫氧化物)交換樹脂,導致序貫轉換,首先轉換為nax鹽且其后轉換為naoh。還公開了用于此類序貫轉換的選擇性滲透膜型轉換器。在轉換后,氫氧化鈉的離子電導率在第二檢測器中進行測量,且與第一檢測器的離子電導率相比較。該論文陳述數據揭示了由于抑制基線中隱藏或與強酸峰重疊的極弱酸的峰。它還陳述這種方法允許估計分析物峰的pk,并且允許無需標準的近似定量。關于該系統的問題包括下述:(1)由于分別在水合氫離子和鈉以及在陽離子和陰離子交換樹脂上的分析物陰離子和氫氧化物之間的離子交換選擇性中的差異,酸形式分析物不完全轉換為naoh;和(2)當來自兩個檢測器的信號求比率時,必須補償離子交換柱中的分析物帶分散。對于弱酸,例如,它可能具有更多的問題,因為存在可用于鈉離子交換的較少游離水合氫離子。
在pct公開wo9418555中,公開了使用ic原理的儀器和方法,其中不同檢測器提供有用的比較信號。具體地,在一種形式的儀器中,通常以色譜樹脂柱形式的分離工具在包含電解質的洗脫劑的存在下分離分析物離子。來自分離工具的流出物流經抑制器工具,用于將電解質轉換為弱離子化形式且將分析物離子轉換為酸或堿形式。抑制流出物流經用于檢測離子種類的電導率的第一檢測器且生成第一信號。系統的該部分是常規抑制的ic。來自第一檢測器的流出物流經鹽轉化器,用于將以酸或堿形式的分析物離子轉換為鹽形式。隨后,分析物的鹽形式的電導率在第二檢測器工具中進行測量且生成第二信號。分析第一信號和第二信號,以代表輸出信號之間的限定關系。
在wo9418555的一個實施例中,以酸或堿形式的分析物離子在具有相反電荷的成鹽離子的單次轉換中轉換為其相應的鹽。例如,對于由"x"代表的分析物陰離子,并且使用na+離子,nax在第二檢測器工具中進行測量。這在本文中被稱為“單次轉換模式”。它公開了使分散降到最低的鹽轉換器,所述分散可使單次轉換類型的峰比率偏斜。一種公開的單次轉換轉換器是在線微電滲析離子源,其通過膜供應成鹽離子。它包括成鹽離子源通道、抑制器流出物流動通道和分隔兩個通道的選擇性滲透離子交換膜。膜包括與成鹽離子相同電荷的可交換離子,并且對離子種類的跨膜通過抗性。電勢施加于離子源通道和抑制器流出物流動通道之間。抑制器流出物流動通道與來自抑制器的流出物流體連通。在操作中,對于酸或堿形式的分析物在第一電導檢測器中生成的信號用關于分析物的鹽形式在第二離子電導檢測器中生成的信號進行評估,以提供非常有用的信息。其他公開的單次轉換轉換器包括離子交換膜屏障的使用,所述離子交換膜屏障不含電解,但具有足以克服donnan屏障的外部酸或堿濃度。另外其他系統包括多孔膜屏障的使用,使用電流或差壓的應用,以驅動酸或堿成鹽離子進入抑制器流出物流動通道內。單次轉換還通過使抑制器流出物流流經離子交換介質例如離子交換樹脂床的柱公開,所述柱具有與分析物離子相反電荷的可交換離子。
wo9418555還公開了其中分析物離子是兩次轉換的“雙重轉換模式”。在這種情況下,分析物離子轉換為(a)與單次轉換模式中相同類型的抗衡離子的鹽,和(b)通過分析物離子的酸或堿形式與所選擇的陰離子和陽離子的同時離子交換,與分析物離子相同電荷的共同單一離子的鹽。在使用選擇性滲透膜的一個實施例中,抑制器流出物在側面為兩個離子源通道的中心通道中流動,所述兩個離子源通道一個包括陰離子并且另一個包括陽離子。選擇性滲透膜使離子源通道與抑制器流出物流動通道隔開,并且包括一類可交換離子,其允許此類陽離子和陰離子輸送進入抑制器流出物流動通道內,以實現雙重轉換。在另一種同時的雙重轉換中,抑制器流出物從第一檢測器流動通過離子交換介質例如離子交換樹脂床,所述離子交換介質包括與選擇性滲透膜中所需相同類型的可交換陰離子和陽離子。還公開了序貫雙重轉換。在一個實施例中,抑制器流出物從第一檢測器序貫流經相反電荷的兩個離子交換柱。例如,第一柱包括與分析物離子相同電荷的共同的單一離子,使得在第一柱中形成具有共同陰離子或陽離子的轉換酸或堿,所述轉換酸或堿傳遞到第二柱用于轉換為鹽,或柱的次序可逆轉。此外,它公開了用于序貫雙重轉換實施例的選擇性滲透膜系統。
在具有逆流流動的化學模式中使用膜抑制器將抑制的色譜流出物轉換為鹽的另一種嘗試公開于yuanhuang,shi-fenmou,ke-naliu,j.chromatography,a832:141-148(1999)中。在這種方法中,僅提供足夠的再生劑溶液,使得抑制不完全。然而,難以控制本底和噪聲。裝置對給定再生劑濃度的再生劑流速和洗脫劑流速兩者非常敏感。
美國專利號4,455,233公開了鹽轉換的另一種方法,使用具有與分析離子相同電荷的共離子的酸或堿的洗脫劑,其中共離子為水合氫離子或氫氧化物形式。在這種方法中,用于陰離子的電解質是酸并且用于陽離子的洗脫劑是堿。洗脫劑和分析物兩者均轉換為鹽形式。盡管洗脫劑在鹽形式中具有的電導率低于導電形式,但這種方法中的本底可高達100us/cm。此類高本底導致更高的色譜噪聲。上述方法一般與用于離子色譜的常用洗脫劑不相容,并且需要容易轉換為更低本底的鹽形式的洗脫劑。
通過karu等人iii的近期綜述解決了通過經修飾的scac朝向弱酸檢測的各種努力;鑒定的最有希望的方法是在常規氫氧化物洗脫劑scac系統后的強堿(例如naoh)引入,隨后為第二電導檢測器(d2)。iv-v在naoh引入后,洗脫酸hx的級分f轉換為nax;f<1并且隨著pka減少而增加(例如,f對于具有pka10的酸將為0.5,如果堿引入后的ph為10)。d2信號因此是負的,并且等于fc(λx--λoh-),其中c是以eq/l表示的洗脫物濃度,并且λx-和λoh-分別為x-和oh-的限制當量電導。該方法對于在scac中響應不明顯的極弱酸(10>pka≥7)是非常有吸引力的。scac信號(d1)和來自d2的信號的組合還可用于鑒定共洗脫,估計pka且執行通用校準。4,5一般方法已擴展,因此它可與標準商購可得的設備一起使用,但極弱酸lod保持在個位數μm水平。vi近來,證實揮發性弱酸洗脫物主要是h2s和hcn(以及co2)可通過非極性膜輸送到堿流內。vii不涉及液體混合,并且噪聲改善≥100x,從而按比例改善lod。這對于構成大多數的非揮發性無機酸,例如硼酸鹽、亞砷酸鹽且特別是硅酸鹽當然不是有效的。
存在將弱解離分析物轉換成鹽形式且有利于針對低本底檢測此類分析物或后續反應產物的用于有效系統的抑制色譜的需要。本發明提供了抑制器、包括抑制器的系統以及使用抑制器和系統滿足該需要的方法。
技術實現要素:
本發明提供了在離子交換色譜、弱解離酸和堿的分離和檢測領域中長期公認但以前未解決的問題的解決方案。本發明提供了用于將揮發性酸或堿引入含有目的分析物的洗脫劑流內的裝置和方法。揮發性酸或堿分別實現作為堿或酸存在的目的分析物的解離。解離的分析物可針對揮發性酸或堿的低本底檢測。在各個實施例中,解離的目的分析物使用電導檢測器進行檢測。
在一個示例性實施例中,本發明提供了用于在含有弱解離酸或堿的混合物的色譜分離后形成弱解離酸或堿的鹽的裝置,所述色譜分離通過使所述混合物經過色譜介質進行。示例性裝置配置用于整合到色譜介質下游的色譜系統內,并且包括浸入揮發性堿或酸的溶液中的可滲透膜。膜采取任何有用的形式(例如平坦或管狀)。可滲透膜允許揮發性堿或酸從溶液通過進入可滲透膜的內腔內,它在其中接觸來自色譜柱的流動流出物,從而將弱離子化的酸或弱離子化的堿轉換為揮發性堿或揮發性酸的相應鹽。
在本發明的另一個方面,鹽轉換在本發明的可滲透膜裝置中執行,利用與可滲透膜的外表面接觸的揮發性酸或堿。酸或堿(或其溶液)可與膜直接接觸,或者膜可與酸或堿蒸氣接觸,例如膜懸浮接近于酸或堿(或其溶液)。本發明的另一個實施例包括用于執行上述方法的系統,所述系統包括(a)在包含與所述分析物離子相反電荷的電解質抗衡離子的洗脫劑的存在下,用于分離所述分析物離子的具有入口和出口的色譜分離器,(b)抑制器,和(c)利用揮發性酸或堿的本發明的可滲透膜裝置。
本發明的一個實施例涉及用于樣品溶液中的分析物離子或多重不同分析物離子的抑制離子分析的方法。分析物離子檢測為在本發明的滲透膜裝置中通過與揮發性堿或揮發性酸反應形成的分析物離子的鹽。該方法包括下述步驟:(a)通過有效分離分析物離子的分離介質以形成分離介質流出物流,用包含與分析物離子相反電荷的電解質抗衡離子的洗脫劑洗脫樣品溶液,(b)使分離介質流出物流流經抑制區,電解質抗衡離子在其中被去除,以將電解質轉換為弱離子化形式,以形成抑制器樣品流出物流,(c)通過與揮發性堿或揮發性酸反應形成分析物鹽流,在滲透膜裝置中將抑制器樣品流出物流中的分析物離子轉換成鹽。其后,檢測分析物鹽。
在本發明的另一個實施例中,鹽轉換在通過與包含揮發性酸或堿的流混合分離后執行,隨后為使用優選的電導檢測器的檢測。在該實施例中,不使用用于引入揮發性酸或堿的膜界面,其中揮發性酸或堿液體直接加入后置柱洗脫劑流中。
本發明的其他實施例、目的和優點由本文提供的詳述將是顯而易見的。
附圖說明
圖1是示意性顯示的本發明的裝置的配置。cr-atc,連續再生的陰離子捕獲柱;paid,滲透性胺引入裝置;d1,第一電導檢測器;d2,毛細管規模的第二電導檢測器;三通管(tee),螺紋10-32和孔尺寸0.50mm。
圖2是示出了使用0.1mmnaoh或0.1mmdea,各種濃度的hcl和hcn(pka9.31)的計算響應的曲線圖。
圖3是示出了膜抑制器的電流對d1和d2處的基線噪聲的作用的曲線圖。噪聲在扣除基線漂移(從數據中扣除的預測的數據最佳線性擬合)后經過2.5分鐘時期進行計算。電流從0.0ma增加到20ma,在每個步驟中為4ma,而koh洗脫劑保持在2.0mm。在d2處的比電導本底為~31μs/cm。在水再循環模式中,在paid裝置后的主要洗脫劑流為再生劑流。在外部水模式中,新鮮的di水流用作再生劑流。再循環模式已使用多天而無噪聲中的增加或者抑制器或cr-atc性能中的劣化。
圖4是示出了使用paid的雙重電導檢測的應用的曲線圖。抑制器在時期0-4和30-36分鐘內以20ma操作,并且在4-30分鐘期間關閉。50μeq/l的10μl注入用于每種離子。來自co2侵入樣品的碳酸鹽和亞硫酸鹽是硫化物標準品中的雜質。頂部跡線是常規抑制的檢測器響應,并且底部兩條跡線是在[dea]的兩個不同水平,27μm和150μm的第二檢測器響應。2mmas11柱;流速0.3ml/分鐘。koh梯度圖形就左縱坐標而言顯示。
圖5是示出了含有下述的硼硅玻璃樣品小瓶中的硅酸鹽的瞬時出現的曲線圖:(a)輕度堿性溶液,(b)di水和(c)微酸性溶液。僅(a)含有起始可檢測濃度的硅酸鹽。樣品在所需時間轉移至聚苯乙烯樣品小瓶。分析條件與圖4中相同。
圖6是示出了使用1.0ml樣品注入,在ag24+as24柱上的雙重電導離子色譜的曲線圖。梯度圖形與圖4相同。抑制器電流20ma0-6和32.5-40分鐘且在6-32.5分鐘期間關閉。樣品濃度(μeq/l):硅酸鹽(2.0),氟化物(0.25)和所有其他離子(1.0)。碳酸鹽非有意添加。在該柱設置下的給定條件下的分離對于硅酸鹽是良好的,但損害其他分析物的分離。丙酮酸鹽、氰化物和硝酸鹽分別與甲酸鹽、硫化物和硫酸鹽共洗脫,并且不存在于該樣品中。
圖7示出了滲透性胺引入裝置(paid)的設計。連接的入口和出口peek管分別長18和10cm。在另一種形式中,teflonaf管纏繞在4mm支撐桿上,通過在100℃下維持30分鐘來熱固化,并且隨后所得到的線圈用于paid中。
圖8是示出了關于在0-200μm范圍內的naoh和二乙胺氫氧化物(deaoh)的理論比電導值的曲線圖。對于dea,實線指示實際數據以及最佳二次擬合,而零截距最佳線性擬合由虛線顯示。
圖9是示出了dea濃度和根據外部dea溶液濃度的流出物比電導的曲線圖。水以0.3ml/分鐘流經浸入所述濃度的dea溶液中的長60cm的teflon
圖10顯示了抑制器電流對第一檢測器(d1)的基線電導率的作用。設置如圖1中。koh洗脫劑濃度保持在相對低的2.0mm,使得抑制器電流自始至終處于大量過量;1ma足以抑制這種洗脫劑。這些顯然過大的電流條件允許檢查系統噪聲的原因,所述系統噪聲也在較低電流下存在,盡管處于較低水平。在典型的抑制器操作中,電流水平在梯度運行期間不變;像這樣,電流在大部分運行期間顯著過量。在paid中,使用18%(v/v)dea溶液,導致31μs/cm的d2本底。洗脫劑流速0.30ml/分鐘;通過d2的流速為paid流出物的20%(0.06ml/分鐘);paid流出物的80%再循環通過抑制器的再生劑通道。電流從0.0ma增加到20ma,步長為4ma。注意當施加的電流朝向洗脫劑抑制時,噪聲一般更低;它是促成噪聲的過量電流。
圖11示出了在不同抑制器電流水平下的負模式電噴霧全范圍質譜;洗脫劑是以0.3ml/分鐘的5mmkoh,抑制器:2mmaers500。2.5ma電流足以抑制這個洗脫劑的量。注意0ma指示對抑制器的功率短暫關閉,它從靜態離子交換容量繼續抑制實質時期。在這種情況下,電流僅在兩個最高電流水平下顯著過量,并且化學噪聲主要在高m/z(>1200)區域中出現。
圖12a示出了兩種不同抑制器的負離子模式esi-ms掃描。洗脫劑是以0.3ml/分鐘的30mmkoh,伴隨50ma的抑制器電流。光譜在負模式和正模式(圖12b)兩者中跨越光譜儀的整個質量范圍(30-1500m/z)進行記錄。負離子的總本底比正離子的那種大得多(圖12b),并且在兩個抑制器之間可比較。單體離子苯乙烯磺酸鹽在m/z183處明確可見(通過碎片光譜證實)。圖12b使用與負離子模式中使用的相同規模,以顯示本底噪聲中的巨大差異。這并不意外,因為抑制器從洗脫劑中去除任何帶正電的干擾物。在圖12a中,苯乙烯磺酸鹽峰是容易鑒定的,并且它作為接枝單體的存在是容易解釋的。然而,它占總離子流的無意義部分,其中許多集中在m/z500-600周圍。
圖12b與圖12a中相同的兩種不同抑制器條件的抑制器的正離子模式esi-ms掃描。縱坐標縮放與圖12a中相同,以示出正模式和負模式之間的本底中的巨大差異。asrs300數據偏移1,5000000個計數,因此兩者可繪制在同一圖上。
圖12c.dionexis25泵直接連接至asrs500,在抑制器前面具有限流器線圈,其以0.5ml/分鐘提供~1200psi的背壓。抑制器在再循環模式下以50ma的電流進行操作。在獲取新的本底光譜之前,使抑制器連續操作大約8天。注意該圖和上一圖之間的縱坐標縮放中的差異。沖洗的確幫助去除特別是對于單體苯乙烯磺酸鹽的一些信號,但對于負模式,本底仍然是令人難以置信的高。在低波長uv吸光度跡線中沒有可與電導噪聲相關的模擬噪聲(下圖13顯示了208nm吸光度跡線)。
圖13抑制器電流對在d1處的基線噪聲的作用(以0.30ml/分鐘的2.0mmkoh)。下跡線,基線電導率跡線;上跡線,在208nm處的agilent1290二極管陣列吸光度跡線。注意電導率噪聲不反映在吸光度數據中,表明光學吸收種類與導電種類不直接相關。在其他實驗中,我們已作出觀察,所述觀察也表明了這一點,例如,在特定的電流變化實驗期間,平均本底電導率可減少,而吸光度增加。
另外,抑制器流出物的uv吸光度僅在極低波長(<205nm)處可測量,光譜是無特征的,其中吸光度在減少的波長處單調上升(圖14),表明負責的物種可能不是芳香族的。
圖14在各種抑制器電流下,關于2mm電生成的koh洗脫劑的抑制的檢測器流出物的吸收光譜。該光譜是無特征的,并且在λ<205nm處顯示一般的指數上升。盡管很可能存在低uv吸收雜質伴隨電流的一般增加,但是它不是特別可再現的,并且吸光度非常低。
圖15在從esrs500抑制器收集并且在5x稀釋后離線測量的各種抑制器電流下的再生劑通道流出物的低波長吸收光譜。洗脫劑是以0.3ml/分鐘的2mmkoh。與圖14中的數據比較;峰吸光度為大約100x大。注意在λ>220nm處沒有觀察到顯著的吸收或光譜特征。還值得注意的是,在該上下文中,即使當不使用抑制器時,一些降解產物也會積累。當抑制器在長期儲存之后首次使用時,uv和質譜研究兩者均指示雜質本底高得多,并且僅在延長的洗滌/使用后才達到穩態值。對于大多數應用,噪聲的增加將不可感知,但在特定應用中,達到穩定狀態可能需要高達一周的連續使用。
圖16a、圖16b、圖16c和圖16d.與圖4相關的校準曲線。(a)在d1處的校準曲線;(b)、(c)、(d)在d2處的校準曲線:關于150μmdea的實心跡線(30.8μs/cm本底)和關于27μmdea的虛線跡線(本底5.5μs/cm)。關于每種離子的濃度范圍為2-200μeq/l。
圖17a和圖17b.在使用150μm和27μm[dea]之間的d2處,各種陰離子的lod比和校準斜率比。檢測極限在表2中列出。本底電導率水平分別為30.8μs/cm和5.5μs/cm,而相應的基線噪聲水平分別為4.4和0.83ns/cm。斜率比1指示靈敏度不變,而值<1指示它對于較低背景[dea](牛磺酸、硅酸鹽、氰化物)較低。lod比落入0.1-0.5的范圍內,對應于從150到27μm[dea]的lod中的10-2x改善。
圖18a、圖18b、圖18c和圖18d.對范圍為(a)強酸至(d)pk=6的單質子洗脫酸的不同濃度(在峰頂點處的50-150μm,d2本底100μmnaoh)的響應。模擬的峰曲線是修正的高斯曲線,并且陰離子的當量電導(λx-)假定為60。w形峰由不足的本底堿濃度產生。轉載自
圖19.使用1.0ml樣品注入的雙重電導檢測。樣品注入時間,0.0分鐘;樣品上樣時間,32.5分鐘;抑制器電流,從6-32.5分鐘的0ma和在剩余時間內的20ma。1.0μeq/l的1.0ml注入用于每種離子。碳酸鹽起因于對樣品的co2侵入。亞硫酸鹽是所使用的硫化物標準品中的雜質。甲酸鹽雜質來自氰化物標準品和來自環境空氣中的甲酸對樣品的侵入兩者。
圖20a、圖20b和圖20c.在d1和d2處的校準曲線。圖20a顯示了基于d1的輸出的校準曲線,并且圖20b和20c顯示了基于d2的輸出的校準曲線。校準范圍對于硅酸鹽為0.04-4.0μeq/l,并且對于所有其他離子為0.02-2.0μeq/l。硅酸鹽、甲酸鹽和氯化物的定量使用as24柱實現,所有其他在as11柱上實現。
圖21a是顯示各種濃度的極弱酸(hcn)的響應(存在來自基線的負信號)的曲線圖。
圖21b是顯示了在100μm堿的背景下的極強酸(hcl)響應的曲線圖,所述堿在堿性中從強堿(pkb<0)到具有pkb=5的堿(接近于氨,pkb4.76)不等。
圖22是顯示了其中酸例如hno3用于滲透膜以使弱酸分析物質子化的實施例的圖解。分析物隨后向下游流動到管道的不同區段,以滲透出管道并且被檢測(例如,使用電導檢測器)。
在附圖的若干視圖自始至終,相同的參考標記指相應的部分。
具體實施方式
引言
本發明的系統可用于測定大量離子種類。本發明提供了用于分離且檢測目的弱酸性或弱堿性分析物的裝置、系統和方法。待測定的種類是酸性或堿性分析物的鹽。合適的樣品包括地表水和其他液體,例如工業化學廢物、體液、飲料和飲用水。本發明的裝置不需要電流源或恒流泵;該設備易于構建和使用。
縮寫
paid是指滲透性胺或酸引入裝置,其是本發明的裝置。如應理解的,本發明的裝置同樣適用于其中它是滲透性酸引入裝置的形式。
定義
當使用術語“離子種類”時,它包括以離子形式和分子組分的種類,所述分子在本發明的條件下是離子化的。
術語“毛細管規模”定義為涵蓋如化學分析中一般使用的窄孔毛細管管道,但并不限于此類毛細管管道。相反,術語“毛細管管道”廣泛地包括尺寸在現有技術毛細管管道的內部尺寸的數量級上的管道。此類毛細管通常具有范圍為約5至約1,000微米,更優選約10至約500微米的孔直徑。此類尺寸任選應用于本發明的滲透性膜裝置、分離器柱或抑制器管道。一段或多段毛細管可連接以形成連續的毛細管管道。毛細管管道導致例如約0.1至約50μl/分鐘的毛細管流速。
“弱解離酸”是具有約5至約10的pka的酸分析物。
“弱解離堿”是具有約5至約10的pkb的堿分析物。
“揮發性酸”或“揮發性堿”指以蒸氣、純凈液體或溶液形式的分別種類。
“可滲透膜”指對離子、氣體、酸和/或堿至少部分可滲透的膜。膜可為任何有用的配置,包括平坦、管狀、多環的等。示例性可滲透膜包括teflonaf和離子交換膜。可滲透膜可為氣體可滲透和液體不可滲透的膜(在給定壓力下)。
“洗脫物”指色譜溶質或分析物。
示例性實施例
本發明提供了在離子交換色譜、弱解離酸和堿的分離和檢測領域中長期公認但以前未解決的問題的解決方案。本發明提供了用于將揮發性酸或堿引入含有目的分析物的洗脫劑流內的裝置和方法。揮發性酸或堿分別實現目的分析物,堿或酸的解離。解離的分析物可針對揮發性酸或堿的低本底檢測。在各個實施例中,解離的目的分析物使用電導檢測器進行檢測。
在一個示例性實施例中,本發明提供了用于在含有弱解離酸或堿的混合物的色譜分離后形成弱解離酸或堿的鹽的裝置,所述色譜分離通過使所述混合物經過色譜介質進行。示例性裝置配置用于整合到色譜介質下游的色譜系統內,并且包括浸入揮發性堿或酸的溶液中的可滲透膜。可滲透膜允許揮發性堿或酸從溶液通過進入可滲透膜的內腔內,它在其中接觸來自色譜柱的流動流出物,從而將弱離子化的酸或弱離子化的堿轉換為揮發性堿或揮發性酸的相應鹽。
本發明的示例性裝置包括中空膜,所述中空膜對以其解離形式的目的分析物基本上不可滲透,并且對揮發性酸或堿可滲透。示例性裝置包括中空可滲透膜,其中洗脫劑流流過中空膜的內腔。膜與揮發性酸或堿接觸。酸或堿滲透膜,從而進入內腔且接觸目的分析物,解離目的分析物,由此形成鹽。目的分析物的鹽形式是可檢測的。
在一個示例性實施例中,揮發性酸或堿引入洗脫物流內。滲透性酸/堿引入裝置包括窄孔膜管,其對揮發性酸或堿是可滲透的,例如teflonaf。teflonaf是由dupont商購可得的無定形氟塑料。酸或堿可滲透膜管浸入揮發性酸/胺中,或置于與這種揮發性組分的溶液緊密接近。scac流出物流過可滲透膜的內腔。二乙胺(dea)由于其低pkb(3.0)和高蒸氣壓而選擇作為胺試劑。本發明的鹽轉換器系統和方法提供了基于低水平的酸或堿引入流出物內的優點。在其中再生劑不是電解再生的一個示例性實施例中,滲透裝置不需要電流源或恒流泵。
在一些實施例中,酸或堿可滲透膜可包括但不限于包含分散在膜各處的親水基團的
在一個示例性實施例中,膜在外部夾套的內腔內,并且揮發性酸或揮發性堿在由膜的外表面和所述外部夾套的內表面形成的環中。揮發性堿或揮發性酸可為流動或靜態的。根據本發明,揮發性酸或堿可為與膜的一側接觸的流體,而scac流出物在另一側上流動。因此,膜可為管狀或平坦配置。
本發明的另一個實施例包括用于執行上述方法的系統,所述系統包括(a)在包含與所述分析物離子相反電荷的電解質抗衡離子的洗脫劑的存在下,用于分離所述分析物離子的具有入口和出口的色譜分離器,(b)抑制器,和(c)利用揮發性酸或堿的本發明的滲透膜裝置。
在一個示例性實施例中,本發明的裝置并入離子色譜系統內,用于分離和檢測弱酸性或弱堿性分析物。離子色譜是用于分析離子的已知技術,其通常包括使用含有電解質的洗脫劑的色譜分離階段和洗脫劑抑制階段,隨后為通常通過電導檢測器的檢測。在色譜分離階段,使用電解質作為洗脫劑從分離柱中洗脫注入樣品的離子。在抑制階段,電解質的電導率而不是分離的離子(如果來源于強酸/堿)的電導率被抑制,使得分離的離子可通過電導池進行測定。這種技術在美國專利號3,897,213;3,920,397;3,925,019;和3,926,559中詳細描述。
圖1顯示了本發明的裝置和并入裝置的系統的示例性配置。在注射器上游,該系統包括水源、koh洗脫劑生成器和泵,以使水和洗脫劑移動通過系統。這種示例性裝置包括連續再生的離子捕獲柱(cr-atc)。cr-atc去除由洗脫劑生成器生成的潛在雜質,并且從thermofisherscientific(dionex,sunnyvale,california)商購可得。在注射器的下游,該系統包括連接到分離柱的保護柱。分離柱進料到抑制器內。離開抑制器的流出物進入第一檢測器,例如電導檢測器。在經過第一檢測器后,流出物經過本發明的滲透性胺或酸引入裝置(paid),其中目的弱解離酸分析物通過滲透通過paid的膜進入抑制流出物流內的揮發性堿或酸轉換為其相應的鹽形式。離開paid的流出物經過三通管的一個臂且進入在其中進行檢測的檢測器,例如電導檢測器。離開paid的流出物任選傳遞通過三通管的另一臂并進入抑制器的再生劑通道內。如應了解的,流出物可同時或序貫地被傳遞到檢測器和抑制器的再生劑通道兩者。
代表性paid裝置顯示于圖1的擴展部分和圖7中。關于抑制器的流出物流入paid裝置的可滲透膜的內腔內。可滲透膜的至少一部分維持在揮發性堿的溶液中。示例性堿是二烷基胺,例如二乙胺。
本領域技術人員應了解,參考圖1闡述的裝置、系統和方法同樣適用于在適當的色譜分離后的弱可離子化堿的檢測。在該實施例中,本發明的可滲透膜裝置維持在揮發性酸的溶液中。揮發性酸穿過與目的分析物接觸的可滲透膜,將所述目的分析物轉換為其鹽形式。
在一個示例性實施例中,圖1的系統的一個或多個部件為毛細管規模。例如,分離柱可為毛細管規模。近來,由于與分離過程的小型化相關的優點,使用內徑1mm或更小的分離柱的毛細管高效液相色譜作為分析分離工具已日益普及。離子色譜中的典型分離柱具有范圍為2mm至4mm的柱內徑,并且以范圍為0.2至3ml/分鐘的流速操作。以毛細管形式(即,使用內徑約1mm或更小的小孔柱)實踐離子色譜潛在具有用于離子分析物分析的許多優點。使用毛細管分離柱可改善分離效率和/或速度。以毛細管形式的分離過程需要量少得多的樣品,且因此提供了與其中樣品量有限的應用的改善的相容性。毛細管離子色譜系統通常以1至20μl/分鐘操作,因此消耗的洗脫劑的量非常小。毛細管離子色譜具有改善的連續操作能力,伴隨最低限度的干預,從而使與系統啟動和關機相關的問題降到最低。在低流速下的毛細管離子色譜操作改善了系統與質譜法的相容性。此外,以毛細管形式的離子色譜實踐打開了使用填充有更奇特和難以制造的固定相或奇特洗脫劑的新柱,對于困難應用提供新的選擇性的可能性的大門,因為很少這樣使用。
在一個示例性實施例中,圖1中顯示的一個或兩個檢測器(d1、d2)為毛細管規模。
圖22顯示了本發明的一個實施例,其中本發明的可滲透膜裝置200分成兩個區,酸引入區段和弱酸提取區段。在酸引入區段中,強酸例如hno3經由酸入口/引入端口220引入膜210的表面。酸滲透膜210且接觸膜210的內部區室內的弱解離酸。過量酸通過酸廢液端口230離開。酸還可直接添加而不是通過膜210添加。弱解離酸穿過酸引入區段的長度,從而進入弱酸提取區段。去離子水經由水引入端口240引入弱酸提取區段內。水接觸膜210的表面,從而提取質子化的弱解離酸并將其攜帶離開可滲透膜裝置200且至檢測器,例如,電導檢測器。
在一些實施例中,可滲透膜裝置200可分成兩個區,堿引入區段和弱堿提取區段。在這些實施例中,在堿引入區段中,強堿經由類似于酸入口/引入端口220的堿入口/引入端口引入膜210的表面。堿滲透膜210且接觸膜210的內部區室內的弱解離堿。堿還可直接添加而不是通過膜210添加。弱解離堿穿過堿引入區段的長度,從而進入弱堿提取區段。去離子水經由水引入端口例如240引入弱堿提取區段內。水接觸膜210的表面,從而提取去質子化的弱解離堿并將其攜帶離開可滲透膜裝置200且至檢測器,例如,電導檢測器。
在一個示例性實施例中,可滲透膜210設置在由材料形成的部件內,所述材料對測定的液體組分基本上不可滲透。不可滲透的外部部件260包括特征在于酸入口端口220和酸出口/廢液端口230的酸引入區段。外部不可滲透部件260還包括與酸引入區段鄰接的弱解離酸提取區段,所述酸引入區段配置具有水入口/引入端口240和弱解離酸出口端口250。在一個示例性實施例中,弱解離酸出口端口250連接到檢測器,例如電導檢測器。可滲透膜210配合在不可滲透部件260的環(或其他腔)內,其中這兩個部件這樣間隔開,使得流體例如酸和水能夠在可滲透膜210的外表面和不可滲透部件260的內表面之間流動。
本發明的示例性系統包括抑制器,其可為在離子色譜中有用或標準的任何形式。抑制離子色譜是用于離子分析的已知技術。電解質的抑制或剝離在美國專利號3,897,213;3,920,397;3,925,019;和3,926,559通過離子交換樹脂床進行描述。不同形式的抑制器柱在美國專利號4,474,664中描述且公開,其中使用以纖維或片材形式的荷電離子交換膜代替樹脂床。在這種形式的抑制器中,樣品和洗脫劑在膜的一側上通過,而在另一側具有流動的再生劑,膜分配來自色譜分離的流出物的再生劑。膜通過與膜的可交換離子相同電荷的離子,以將洗脫劑的電解質轉換成弱離子化形式,隨后為離子的檢測。
另一種膜抑制器裝置公開于美國專利號4,751,004中。其中,中空纖維抑制器填充有聚合物珠,以減少帶擴展。存在此類填充可與其他膜形式一起使用的建議。此外,存在通過使用離子交換填充珠來改善纖維抑制器的功能的建議。沒有闡述關于這種顆粒為何以改善方式起作用的理論。
另一種抑制系統公開于美國專利號4,459,357中。其中,來自色譜柱的流出物經過由通道兩側上的平坦膜限定的開放流動通道。在兩個膜的相對側上是再生劑溶液經過其的開放通道。與纖維抑制器一樣,平坦膜傳遞與膜的可交換離子相同電荷的離子。電場在流出物通道的相對側上的電極之間通過,以增加離子交換的移動性。關于這種電滲析膜抑制器系統的一個問題是需要非常高的電壓(50-500伏dc)。當液體流變為去離子時,電阻增加,導致大量的熱產生。這種熱對于有效檢測是有害的,因為它極大增加了噪聲且降低了靈敏度。
在美國專利號4,403,039中,公開了另一種形式的電滲析抑制器,其中離子交換膜采取同心管的形式。電極之一在最內管的中心。關于這種形式的抑制器的一個問題是有限的交換容量。盡管電場增強了離子遷移率,但該裝置仍依賴于本體溶液中的離子向膜的擴散。還參見美國專利4,500,430和4,647,380。
另一種形式的抑制器在美國專利號4,999,098中描述。在這種儀器中,抑制器包括由離子交換膜片隔開的至少一個再生劑區室和一個色譜流出物區室。該片材允許與其可交換離子相同電荷的離子的跨膜通過。離子交換篩用于再生劑和流出物區室。來自流出物區室的流動被導向檢測器,例如電導率檢測器,用于檢測分辨的離子種類。篩提供離子交換部位且作用于提供跨越流出物流動通道的位點至位點轉移路徑,使得抑制能力不再受本體溶液中的離子擴散到膜的限制。還公開了夾心抑制器,其包括與第一膜片相對且限定第二再生劑區室的第二膜片。公開了沿抑制器的長度與兩個再生劑室連通的間隔開的電極。通過跨越電極應用電勢,存在裝置的抑制能力中的增加。該專利公開了在再生劑流動通道中流動且由再生劑遞送源供應的通常再生劑溶液(酸或堿)。在通常的陰離子分析系統中,氫氧化鈉是電解質顯影劑且硫酸是再生劑。該專利還公開了使用水來替換電滲析模式中的再生劑溶液的可能性。
本發明的示例性系統包括一個或多個洗脫劑生成器。美國專利號6,036,921和美國專利號6,225,129描述了通過使用水作為載體可用于生成高純度酸和堿溶液的電解裝置。使用這些裝置,在線自動生成高純度、無污染的酸或堿溶液,用作色譜分離中的洗脫劑。這些裝置簡化了梯度分離,所述梯度分離現在可使用具有最小延遲的電流梯度代替使用常規機械梯度泵執行。示例性洗脫劑生成器在美國專利號8,647,576中描述。通過下述步驟在水性溶液中生成酸或堿:(a)在第一酸或堿生成區中,提供通過第一屏障(例如,陰離子交換膜)隔開的與水性液體鄰近的第一離子源,所述第一屏障基本上防止液體流動并且僅輸送與所述第一離子相同電荷的離子,(b)在第二酸或堿生成區中,提供通過第二屏障隔開的與水性液體鄰近的相反電荷的第二離子源,所述第二屏障僅輸送與第二離子相同電荷的離子,以及(c)通過施加電勢通過所述第一區和第二區跨越第一屏障輸送離子,以在所述第一區或第二區之一中生成含酸水性溶液以及在另一區中生成含堿水性溶液,所述含酸水性溶液和含堿水性溶液可結合以形成鹽。
本發明的示例性系統包括一個或多個檢測器。在離子色譜中,基于分析物的性質選擇特定的檢測方案。例如,硝酸鹽、溴化物或碘化物的分析可以通過紫外線檢測(uv)來進行,因為這些分析物在uv中吸收。然而,其他常見離子如氟化物、硫酸鹽和磷酸鹽不吸收uv,因此不響應直接uv檢測。
在各個實施例中,系統包括一個或多個電導檢測器。電導檢測是本體特性檢測,并且總電導取決于通過離子上的電荷的離子性質以及樣品中的遷移率和濃度。溶液的比電導是存在的不同離子的濃度-遷移率乘積的總和。眾所周知等濃度的特定不同化合物,例如nacl和hcl,具有非常不同的比電導。然而,電導率響應于所有離子溶質,但無法提供總電荷的測量。
根據本發明,示例性揮發性胺是能夠從揮發性胺的溶液滲透通過本發明的滲透膜裝置的可滲透膜的胺,膜在所述揮發性胺的溶液中接觸,而含有一種或多種弱可離子化的酸的洗脫劑流在可滲透膜的環內流動。在本發明的paid中使用的示例性揮發性胺包括具有根據式i的一般結構的那些:
其中r1、r2和r3選自h以及取代或未被取代的烷基。示例性烷基部分包括c1、c2、c3、c4、c5、c6、c7和c8直鏈、支鏈和環狀的取代或未被取代的烷基部分。在各個實施例中,r1、r2和r3中的兩個或更多個連同它們與之鍵合的氮一起連接以形成環結構。胺可個別使用或組合使用。
示例性揮發性胺包括但不限于(甲基)n胺、(乙基)n、(丙基)n胺和(丁基)n胺,其中n為1、2或3;烷醇胺(包括但不必限于單乙醇胺(mea)、甲基二乙醇胺(mdea)、二乙醇胺(dea));乙二胺(eda)、甲氧基丙胺(mopa)、二乙基氨基乙醇(deae)等等及其混合物。盡管氨嚴格來說不是胺,但在本文上下文中,氨被包括在與胺相同的氮化合物組中。
在各個實施例中,有用的胺包括具有約10.5至約12的pka的相對更強的胺。在一個非限制性實施例中,胺不含氧。在另一個非限制性實施例中,胺是具有約10.7至約11.4的pka范圍的二烷基胺。在各個實施例中,胺具有小于約95℃,例如小于約70℃、小于約50℃或小于約40℃的正常沸點。合適的胺包括但不限于二甲胺、二乙胺、二丙胺、二異丙胺、二正丁胺、二異丁胺、二仲丁胺、二叔丁胺、吡咯烷、哌啶及其組合(例如混合物)。
在本發明中使用的示例性揮發性胺在所使用的濃度下具有足夠低的電導,使得它們不會過度干擾分析物鹽的檢測。示例性胺是二乙胺。圖8是示出了關于在0-200μm范圍內的naoh和deaoh的理論比電導值的曲線圖。對于dea,實線指示實際數據以及最佳二次擬合,而零截距最佳線性擬合由虛線顯示。圖9是示出了dea濃度和根據外部dea溶液濃度的流出物比電導的曲線圖。水以0.3ml/分鐘流經浸入所述濃度的dea溶液中的長60cm的teflon
本發明的裝置還可用于分離弱堿和用揮發性酸檢測其鹽。在該實施例中,本發明的裝置這樣配置,使得類似于paid裝置的揮發性堿,揮發性酸(或其溶液)在可滲透膜外部。
根據本發明,示例性揮發性酸是能夠從揮發性酸的溶液滲透通過本發明的滲透膜裝置的可滲透膜的酸,所述揮發性酸的溶液與膜接觸,而含有一種或多種弱可離子化的酸的洗脫劑流在可滲透膜的環內流動。有用的揮發性酸包括具有<0至約4的pka范圍的那些酸。揮發性酸的另一個定義是在真空下相對容易地從溶液中去除的酸。示例性揮發性酸包括所有鹵代酸(例如hf、hcl、hbr和hi)以及甲磺酸、甲苯磺酸、羧酸如甲酸等,以及在真空(例如,<10mmhg壓力)下在24小時內至少99%可被去除的其他酸。
任何有用濃度的堿或酸可并入本發明的可滲透膜裝置中。在一個示例性實施例中,滲透溶液中的揮發性胺濃度為約100μm或更低。
在一個示例性實施例中,本發明使用在膜外部的高濃度的揮發性堿或揮發性酸,并且堿或酸中等可滲透通過膜。以這種方式,達到膜內所需的濃度。揮發性堿或揮發性酸的高外部濃度與這些組分跨越膜的適度滲透性確保了外部堿或酸僅需要很少補充。“高外部濃度”意指揮發性堿或酸的濃度足夠高,以至于不需要揮發性堿或揮發性酸的不斷改變、替換或補充。
假定裝置具有短長度(5-75cm)的胺引入管道,示例性外部揮發性堿或揮發性酸濃度足以提供范圍為約100μm至約200μm,且更優選約20μm至約100μm的在膜內的堿或酸濃度。盡管根據某些范圍進行敘述,但應理解包括從最低下限到最高上限的所有范圍,包括在該全范圍或任何具體敘述的范圍內的所有中間范圍或特定值。在普通技術人員的能力范圍內的是,選擇膜滲透性(例如截留分子量、長度、組成、厚度等)以及堿或酸的濃度和特性以實現在膜內的所需堿或酸濃度。在一個示例性實施例中,該裝置能夠延長使用,而無需替換或補充揮發性堿或酸。
在各個實施例中,本發明提供了生成具有低水平的基線噪聲的色譜圖的系統。例如,圖3是示出了膜抑制器的電流對d1和d2處的基線噪聲的作用的曲線圖。噪聲在扣除基線漂移(從數據中扣除的預測的數據最佳線性擬合)后經過2.5分鐘時期進行計算。電流從0.0ma增加到20ma,在每個步驟中為4ma,而koh洗脫劑保持在2.0mm。在d2處的比電導本底為~31μs/cm。在水再循環模式中,在paid裝置后的主要洗脫劑流用作再生劑流。在外部水模式中,新鮮的di水流用作再生劑流。在該實施例中,再循環模式可使用多天而無噪聲中的增加或者抑制器或cr-atc性能中的劣化。
在各個實施例中,本發明提供了具有至少兩個檢測器的系統。代表性系統包括兩個電導檢測器。圖4示出了使用paid的雙重電導檢測的應用。抑制器在時期0-4和30-36分鐘內以20ma操作,并且在4-30分鐘期間關閉。50μeq/l的10μl注入用于每種離子。來自co2侵入樣品的碳酸鹽和亞硫酸鹽是硫化物標準品中的雜質。頂部跡線是常規抑制的檢測器響應,并且底部兩條跡線是在[dea]的兩個不同水平,27μm和150μm的第二檢測器響應(paid后)。2mmas11柱;流速0.3ml/分鐘。koh梯度圖形就左縱坐標而言顯示。
樣品雙重電導色譜圖顯示于圖19中。雙重檢測使用1.0ml樣品注入來實現。樣品注入時間,0.0分鐘;樣品上樣時間,32.5分鐘;抑制器電流,從6-32.5分鐘的0ma和在剩余時間內的20ma。1.0μeq/l的1.0ml注入用于每種離子。碳酸鹽起因于對樣品的co2侵入。亞硫酸鹽是所使用的硫化物標準品中的雜質。甲酸鹽雜質來自氰化物標準品和空氣中的甲酸鹽對樣品的侵入兩者。
在各個實施例中,本發明提供了用于檢測樣品中的硅酸鹽的裝置、系統和方法。圖5是示出了含有下述的硼硅玻璃樣品小瓶中的硅酸鹽的瞬時出現的曲線圖:(a)輕度堿性溶液,(b)di水和(c)微酸性溶液。僅(a)含有起始可檢測濃度的硅酸鹽。分析條件與圖4中相同。
本發明的方法提供了硅酸鹽與其他離子的良好分離。圖6是示出了使用1.0ml樣品注入,在ag24+as24柱(由thermofisherscientific,dionex,sunnyvale,california,u.s.a.商購可得的陰離子交換保護柱和分析柱)上的雙重電導離子色譜的曲線圖。
本發明的裝置提供了跨越不同分析物結構在一系列分析物濃度上的良好線性應答。圖16a-16d是與圖4相關的校準曲線。圖16a顯示了在d1處的校準曲線,并且圖16b-16d顯示了在d2處的校準曲線:關于150μmdea的實心跡線(30.8μs/cm本底)和關于27μmdea的虛線跡線(本底5.5μs/cm)。關于每種離子的濃度范圍為2-200μeq/l。圖17a和17b提供了在使用150μm和27μm[dea]之間的d2處的lod比和校準斜率比,以及各種陰離子的lod比。檢測極限在表2中列出。本底電導率水平分別為30.8μs/cm和5.5μs/cm,而相應的基線噪聲水平分別為4.4和0.83ns/cm。斜率比1指示靈敏度不變,而值<1指示它對于較低背景[dea](牛磺酸、硅酸鹽、氰化物)較低。lod比落入0.1-0.5的范圍內,對應于從150到27μm[dea]的lod中的10-2x改善。
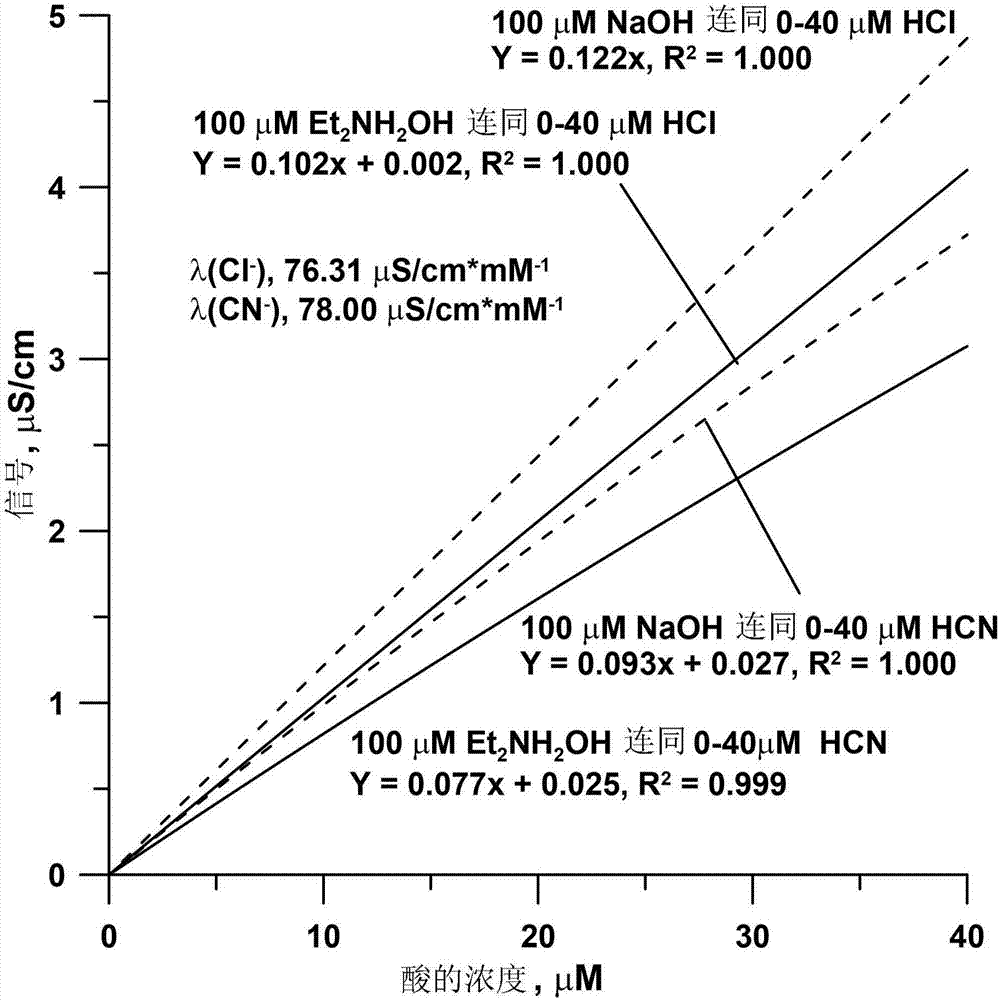
本發明的裝置作用于允許檢測弱離子化分析物的鹽。圖21a是顯示各種濃度的極弱酸(hcn)的響應(這些是來自基線的負信號)的曲線圖。圖21b是顯示了在100μm堿的背景下的極強酸(hcl)響應的曲線圖,所述堿在堿性中從強堿(pkb<0)到具有pkb=5的堿(接近于氨,pkb4.76)不等。圖2是示出了使用0.1mmnaoh或0.1mmdea,各種濃度的hcl和hcn(pka9.31)的計算響應的曲線圖。
利用本發明的裝置的系統和方法對分析物鹽的濃度敏感。圖18a-18d提供了對范圍為(a)強酸至(d)pk=6的單質子洗脫酸的不同濃度(在峰頂點處的50-150μm,d2本底100μmnaoh)的示例性檢測器響應。模擬的峰曲線是修正的高斯曲線,并且假定為60。w形峰由不足的本底堿濃度產生。轉載自
本發明的裝置、系統和方法還允許定量檢測的分析物鹽。圖20a-20c是在d1和d2處的校準曲線。(a)在d1處的校準曲線;(b)、(c)在d2處的校準曲線。校準范圍對于硅酸鹽為0.04-4.0μeq/l,并且對于所有其他離子為0.02-2.0μeq/l。硅酸鹽、甲酸鹽和氯化物的定量使用as24柱實現,所有其他在as11柱上實現。
本發明還提供了使用并入本發明的裝置的系統分離且檢測目的分析物的方法。
本發明的一個實施例涉及用于樣品溶液中的分析物離子或多重不同分析物離子的抑制離子分析的方法。分析物離子檢測為在本發明的滲透膜裝置中通過與揮發性堿或揮發性酸反應形成的分析物離子的鹽。該方法包括下述步驟:(a)通過有效分離分析物離子的分離介質以形成分離介質流出物流,用包含與分析物離子相反電荷的電解質抗衡離子的洗脫劑洗脫樣品溶液,(b)使分離介質流出物流流經抑制區,電解質抗衡離子在其中被去除,以將電解質轉換為弱離子化形式,以形成抑制器樣品流出物流,(c)通過與揮發性堿或揮發性酸反應形成分析物鹽流,在滲透膜裝置中將抑制器樣品流出物流中的分析物離子轉換成鹽。其后,檢測分析物鹽。
在一個示例性實施例中,本發明的裝置不伴隨抑制器的使用而使用。例如,其為弱解離酸或弱解離堿的分析物在色譜過程中與混合物的其他組分分開,并且隨后經過本發明的裝置,分析物在其中離子化,并且通過本發明的裝置下游的檢測器檢測。在一個示例性過程中,分析物使用水進行色譜,由此消除在系統中的抑制器的需要。
本發明的示例性方法提供了與其中不采用本發明的滲透膜裝置的方法相比較的改善的檢測。代表性方法對于酸或堿的鹽由檢測器產生的信號的量級大于在不存在滲透膜裝置的情況下以相同方法對于酸或堿產生的相應信號。
本發明的裝置可用于包括不同類型檢測器的色譜系統中。在一個實施例中,本發明的系統并入除電導檢測器外的檢測器。
下述實例預期進一步示出本發明的所選實施例,并且不應解釋為限制本發明的范圍。
實例
實例1
實驗部分
下文不另行說明的所有設備/部件均來自www.thermoscientific.com。使用具有一個分析通道(2-4mmφ柱)和一個毛細管通道(0.4mmφ柱)的ics-5000離子色譜(ic)(圖1)。色譜在35℃下在ag11+as11或ag24+as24柱(2mmφ)上,使用以0.30ml/分鐘的koh洗脫劑梯度的2mmers-500膜抑制器和10μl或1.0ml樣品體積執行。重要的是,在一些應用中,電滲析膜抑制器在色譜期間關閉所需時期(參見下文,在所使用的洗脫劑濃度時,抑制器的靜態離子交換容量是這樣的,使得即使在關閉后,它仍可維持>30分鐘的抑制)。胺通過paid(浸入水性dea溶液中的teflon
結果與討論
滲透性堿引入和堿的選擇
對弱酸檢測的堿引入的過去努力已涉及強堿。離子化種類無法通過氣體可滲透膜容易地滲透引入。揮發性的未離子化分子可更容易地經過氣體可滲透膜。二乙胺(dea)是一種折衷方案:具有3.0的pkb,顯著量在含有百分比水平的dea的水性溶液中未離子化,并且它不是特別有氣味或有毒的。deaoh的無限稀釋當量電導(240μs(cm*mm)-1)僅略低于naoh的那種(248μs(cm*mm)-1)。在本專利申請中有利的濃度范圍(0-0.2mm)內,naoh預期具有線性濃度-比電導關系;對于dea理想地遵循二次關系(sp.cond,μs/cm=–1.47x10-4c2+0.234c+0.044,r2=1.0000),但是在該有限范圍內,零截距線性擬合也是可接受的(sp.cond,μs/cm=0.2103c,r20.9982),圖8),由于不完全離子化,斜率比naoh的斜率低15%。對于強酸分析物hcl和弱酸分析物hcn(pka9.3)直到40μm的峰濃度(考慮到典型的色譜稀釋,這對應于對于大多數離子>10ppm的注入濃度),預期信號(峰高度)容易計算并且顯示于圖2中。naoh和dea兩者均提供了線性響應,但是對于dea,絕對響應可預測地比naoh低~20%。然而,如果這種靈敏度損失通過缺乏稀釋和通過滲透引入的更好混合得到補足,則這將比過去的方法有吸引力得多,尤其是既不需要泵也不需要電學工具來引入堿。
滲透的dea濃度的關系
滲透的dea的量預期與外部溶液中未離子化的dea的濃度成比例。在目的范圍內,外部溶液中的dea濃度相對很高,因此其大部分是未離子化的,并且未離子化的濃度因此與總濃度線性相關。結果,發現滲透的dea濃度與外部濃度成線性比例(圖9)。注意與外部存在的相比較,滲透的dea的量非常小,進料可使用非常長的時期。對于在15℃下含有18%dea的125ml外部溶液和0.3ml/分鐘的內部流速,系統將在外部濃度中存在1%相對降低之前操作>800小時。該操作時期可通過緩沖系統以具有少量的強酸甚至更延長,使得甚至必須進一步提高外部總dea濃度以維持與之前相同的游離[dea]。然而,由于再填充/濃度調整之前所需的操作時期已經非常長,所以未探究這一點。
在電滲析抑制中的降解產物。抑制器流出物的表征
化學抑制可在scac中提供相對低的噪聲水平,并且在電滲析抑制中,基線噪聲水平可隨著抑制器電流增加超過抑制所需的最小閾值而增加。viii盡管自從這些早期觀察以來,總噪聲水平已降低超過兩個數量級,但使用本文的電滲析抑制器可觀察到相同現象。這種噪聲的確切原因從未得到確定。如果在電解期間的膜降解產生一些極弱酸,則其濃度的瞬時變化將產生噪聲。然而,由于是極弱酸,這種噪聲在堿添加后將極大放大。
圖10顯示了伴隨可見的抑制器電流大量過量生成的基線噪聲。噪聲更可能來自膜的氧化還原降解ix而不是微泡。
所有觀察均與過量的抑制器電流一致,導致其為極弱酸性的較高質量(聚合物/低聚物)降解產物,導致在堿引入后在d2處顯著更大的噪聲。如果在電極/膜界面處的過程負責導致觀察到的噪聲的產物,則察看來自抑制器的再生劑通道流出物可提供更好的洞察。實際上,再生劑流出物的光譜檢查顯示低uv中的吸光度隨著抑制器電流的增加而增加,絕對值是中央通道流出物的那些的~100倍大(圖15)。
在本文設置中,一些噪聲還可源于存在于液體中的dea的可能回滲/氧化,所述液體再循環通過再生劑通道。圖3的確顯示噪聲明顯較小,尤其是對于d2,如果淡水而不是paid流出物為再生劑。
使用化學或間歇電滲析抑制相對于連續電滲析抑制
電滲析膜降解相關噪聲顯然可通過使用化學抑制來消除。大多數(但不是全部)電滲析抑制器也可化學再生,尤其是如果待抑制的洗脫劑濃度不是特別高時。間歇電滲析操作基本上導致在進行色譜分析時間期間的化學抑制。如果可維持完全的洗脫劑抑制,則這代表了兩全其美,以樣品流通量為一定代價。本文的esrs500抑制器具有足夠大的靜態(未施加功率)離子交換容量,以完全抑制以0.3ml/分鐘的10mmkoh共30分鐘。在樣品注入時關閉抑制器的電源并且在完全分離后重新開啟提供了最佳的基線噪聲。還能夠在特定區域中打開或關閉抑制,雖然基線和短暫的峰/傾角中存在偏移(參見圖13),平衡是快速的,并且如果不存在峰/傾角附近的目的分析物峰,則可進行實踐。電解抑制還生成氧;我們已觀察到對于連續電滲析抑制的氧化敏感性硫化物的嚴重損失。8scac先前無法在痕量水平下檢測這種弱酸;為了利用新發現的能力,預防這種損失需要適當的抑制技術。
內部體積、分散和基線噪聲
裝置的幾何形狀和內部體積控制帶分散。給定相同的幾何形狀,較大的停留體積通常以更大的分散為代價改善混合并降低噪聲。提供了在類似努力中的以前報告的性能數據,其中最小的分散和噪聲水平為78±4μl(25μl注入樣品)和5±2ns/cm。然而,在所有以前的情況下,在堿引入之后使用混合線圈或等價物,并且在上文中不考慮由該附加裝置引起的分散。對于上述的最低噪聲情況,總分散包括由混合裝置產生的分散為132±4μl。paid誘導的分散使用10μl無需柱注入的80μmhno3樣品操作測量為48.8±0.2μl。在不存在電滲析抑制的情況下,無論淡水還是paid流出物用于抑制器再生,在基線噪音方面不存在統計學差異,參見圖3中對于0ma電流的d2數據),因此使用流出物再循環。在不存在抑制器電流的情況下,關于30.8μs/cm的本底電導的基線噪聲為4.4±0.6ns/cm,值得注意的是不需要在許多先前研究中使用的進一步的混合線圈。總體上,這種性能至少等于或優于先前的方法(參見表1)。5,6,x
然而,線性配置(或大線圈半徑)據報道最易于混合不良和大分散,xi因此我們將paid中的teflonaf管纏繞在3.7mm支撐桿上,并通過將其放在沸水中30分鐘使形狀熱固化。在相同的測試條件下,噪聲和分散兩者分別降低至3.6±0.2ns/cm和30.3±0.3μl。這種改善在工作的后期發現;本論文中報道的其他數據均無需纏繞而獲得。
通過減少本底改善檢測極限(減少添加的堿濃度)
最常見的是,基線噪聲可與本底的絕對值直接關聯。xii減少引入的堿濃度因此預期隨著本底電導而改善檢測極限(lod),并且因此本底噪聲將降低。雖然較低的堿濃度還可限制測量上限,但當lod改善是主要目標時,這一點較不重要。此外,測量上限可無需用較低的堿濃度來犧牲,參見下文。
從樣品注入到檢測在峰頂點處的典型稀釋因子為~10;因此10μm堿足以測量高達100μm的單質子酸hx(總計例如3.5mg/l氯化物至10mg/l高氯酸鹽),是用于痕量分析的顯著量。然而,對于極弱酸,降低引入的堿量降低了ph,并且由于不充分的離子化可降低信號。對于強酸如hcl,使用強堿引入,信號保持恒定并且與堿濃度無關(直到存在的堿量不足以中和hcl)。使用dea,較弱的堿,信號實際上隨著[dea]的增加而減少,因為形成了緩沖液。然而,作為第一近似,如果噪聲與本底成比例,則s/n比(snr)隨著[堿]增加,與[堿]-1成線性比例,導致在naoh和dea引入之間沒有snr差異。hcn的預期行為在質量上相似;10μmhcn信號保持幾乎相同,降至50μm[堿],但其后由于不完全離子化而急劇下降。隨著[堿]的減少,snr的總增益不如hcl的陡峭,但此處snr也隨著[堿]的減少而一致地增加。從200到10μmdea,關于10μmhcl和hcn的snr增益分別為21.7和6.8x。同樣重要的是,在naoh相對于dea引入之間對于弱酸如hcn預測不出可辨別的snr差異。
然而;可能存在除與本底相關的噪聲源外的噪聲源,因此噪聲可能隨本底電導停止線性減少。此外,計算忽略了不可避免的co2侵入。圖4顯示了含有各種陰離子的樣品的雙重檢測色譜圖;示出了關于兩個不同[dea]值的d2軌跡。可預測地,兩性離子牛磺酸和極弱酸陰離子硅酸鹽/硫化物/氰化物在d1處產生可忽略或不存在的響應,但在d2處產生良好的響應。27μm相對于150μm[dea]本底的d2響應遵循理論預測的行為。強酸信號在較低的[dea]下增加,但是對于極弱酸,信號隨著[dea]的減少而減少。[dea]從150到27μm的5.6x減少幾乎確切地反映在噪聲從4.4到0.83ns/cm的5.4x減少中。因此,盡管在較低[dea]下的響應降低,lod對于極弱酸仍改善。圖16a-16d描繪了在2-200μm范圍內,在圖4中存在的所有注入離子的非常良好的線性校準曲線。圖17a和17b分別概括了隨著[dea]從150降低到27μm,lod的改善和每種離子的靈敏度(校準斜率)變化;所有lod降低,除3個之外的所有靈敏度改善。lod在表2中列出,對于極弱酸分析物的亞μmlod以前從未用電導檢測獲得。
在高分析物濃度下在第二檢測器處的定量
在高分析物濃度下,峰洗脫酸濃度可超過引入的堿濃度。雖然顯然這樣的樣品可被稀釋且再注入,但良好的定量實際上由現存響應是可能的。對于強酸分析物,直觀的是在這種情況下,響應將是w形峰;當所有堿被中和時,信號最初下降并達到最小值,隨后當加入過量的酸時,信號再次開始上升(參見圖18)。取決于酸過量的程度及其pka,w峰的中心可超過原始基線。直觀的是在任何給定高度處(不是峰最大值的部分而是在固定縱坐標值處)的峰寬度必須是分析物濃度的函數。對于標準差s、幅度a的高斯峰,容易得出在任何高度h處的寬度wh由下式給出:
wh=2s(2lna/h)0.5…(1)
類似的關系幾乎適用于任何色譜峰形,除了指數0.5不同之外。峰因此可基于寬度進行定量,只要在檢測器響應中的任何異常(由于不足的[堿]的飽和、檢測器飽和/非線性等)發生之前選擇h,如圖18的左上象限示出的。目前的關鍵點在于定量可在加入的堿完全中和之后很長時間內繼續完成。由于可使用當今使用的快速數據采集系統以高瞬時分辨率測量寬度,僅添加少量的堿對于本文系統和相似系統是足夠的。
重要的paid應用。硅酸鹽的測定
硅在最豐富的地殼元素中;所有天然水中均含有一些溶解的二氧化硅。天然水中的二氧化硅含量通常在100-500μm(2.8-14mg/lsi)的范圍內。xiii美國測試和材料學會(americansocietyfortestingandmaterials)規定了關于1型、2型和3型試劑級別水的最大二氧化硅水平分別為50nm、50nm和8.3μm。xiv溶解的二氧化硅在許多領域中是有問題的。在發電廠鍋爐給水中,它腐蝕加熱設備和渦輪機并降低渦輪機效率。xv在較低nm水平下的測量是半導體工業中需要的,因為亞微摩爾水平仍可影響硅晶片上的表面反應xvi,且低水平的溶解二氧化硅不能通過電導率或碳測量儀器檢測。硅酸鹽也是必需的水生大量營養素。在測量硅酸鹽的許多方法中,xvii在酸性介質中與鉬鹽反應以形成黃色硅鉬雜多酸或其還原產物雜多藍,xviii,xix并且它們的分光光度測量是最常見的。這對于一些樣品是令人滿意的,xx但當痕量必須被測量和/或基質是復雜的時,則不是。離子排斥色譜(ice)和/或電感耦合等離子體質譜(icp-ms)對硅酸鹽/硅測定的首次應用xxi獲得80nm的檢測極限(lod);這仍是通過icp-ms報告的最佳lod。有趣的是,這些作者報告了對于石英炬比氧化鋁炬更低的硅空白信號。在任何情況下,這種方法都是資本密集型的,需要熟練的操作者,并且仍然不能滿足半導體工業的需求。li和chenxxii報道了硅酸鹽的ice分離和電導檢測,僅用水作為洗脫劑,具有20nm的lod。不幸的是,這份報告是不可信的。雖然它已經被引用了33次,但沒有明顯的嘗試來復制結果。在未能復制結果之后,我們認識到這種方法可顯示為從頭開始以降低缺少所要求的lod的數量級。
本文方法足夠靈敏以容易地測定即使在酸性條件下保持在玻璃小瓶中的初始純水中硅酸鹽的逐漸出現,如圖5中所示。如可預料的,硅酸鹽在堿性溶液中在濃度中基本上線性繼續增加。有趣的是,硅酸鹽在純水中的初始出現甚至更快(這種觀察是可再現的)。
盡管比先前報道的方法更敏感,但1.0μm的上述硅酸鹽lod仍不足以用于測試i型試劑水中的順應性。因此,調查大體積樣品注入以滿足該目標。雖然對于使用的ag11-as11柱獲得良好的雙重檢測色譜圖(圖19),但我們發現樣品/水暴露于我們的實驗室環境導致在這些條件下與硅酸鹽共洗脫的痕量甲酸鹽污染。使用具有完全不同選擇性的ag24-as24柱組實現了更穩健的方法。使用相同的koh梯度,硅酸鹽首先洗脫并與所有其他陰離子充分分離(圖6)。因此實現的硅酸鹽的lod基于s/n=3標準為21nm,足以測定對1型試劑水規格的順應性。1ml注入體積還分別允許對于牛磺酸、硫化物和氰化物的3、3和13nmlod。我們希望注意,圖6中的分離沒有針對硅酸鹽的分離最佳化,所述硅酸鹽實際上在此以低效率洗脫。毫無疑問,在許多樣品中,組合物將允許較早洗脫,隨著更尖銳的峰具有更陡的梯度,從而進一步改善lod。在本文洗脫條件下不同分析物的校準曲線顯示于圖20a-20c中。
paid是穩健的低分散、低噪聲裝置,其為雙重電導檢測帶來顯著的簡單性和易用性,以改善弱離子化分析物的可檢測性。測量低水平硅酸鹽的能力是可特別有用的屬性。雖然此處未例證,但顯而易見酸可引入,正如對于極弱堿例如各種胺檢測一樣容易。
實例2
paid的構建
長度60cm的teflonaf管(0.28mmi.d.x0.68mmo.d.,www.biogeneral.com)用于氣態胺滲透,如圖7中所示。將每個端部插入聚四氟乙烯(ptfe)管套筒(25mmx0.71mmi.d.)內,并且通過標準壓縮配合密封到peek接頭(10-32螺紋,0.25mm孔)。將接頭的另一端部連接到peek管道(0.25mmi.d.),其插入穿過在125ml錐形瓶上緊緊密封的硅塞子。燒瓶含有100ml3.0-18%(v/v)dea溶液。抑制器和paid均維持在外殼中在15℃下。
分散測量
為了測量由于paid的分散,將該裝置放在d1之前,在外部具有dea。分散測量為帶體積的平方之間的差的平方根((w2-w’2)1/2,其中w和w’分別是伴隨和不伴隨paid存在的帶體積(參見anal.chem.1984,56,103-105)。
應理解本文所述的實例和實施例僅用于舉例說明性目的,并且本領域技術人員將想到根據其的各種修改或改變,并且這些修改或改變包括在本專利申請的精神和范圍以及所附權利要求的范圍內。出于所有目的,本文引用的所有出版物、序列登錄號、專利和專利申請以引用的方式在此全文并入。
表1.目前和先前研究中的分散體積和基線噪聲。
表2.在d1和d2處的檢測極限(lod)。a
a校準范圍對于150μmdea引入為10-200μeq/l,并且對于27μmdea引入為2-200μeq/l。對于1.0ml樣品注入,校準范圍對于硅酸鹽為0.04-4.0μeq/l,并且對于所有其他離子為0.02-2.0μeq/l。
b丙酸鹽不添加用于1ml樣品注入,由于與硅酸鹽的一定共洗脫。
c具有1ml樣品大小的硅酸鹽(以及甲酸鹽和氯化物)的lod使用as24柱進行計算。
[i]vanatta,l.e.trac,trendsanal.chem.2001,20,336-345.
[ii]michalski,r.mini-rev.med.chem.2014,14,862-872.
[iii]karu,n.;dicinoski,g.w.;haddad,p.r.trac,trendsanal.chem.2012,40,119-132.
[iv]berglund,i.;dasgupta,p.k.;lopez,j.l.;nara,o.anal.chem.1993,65,1192-1198.
[v]
[vi]liao,h.;dasgupta,p.k.;srinivasan,k.;liu,y.anal.chem.2015,87,793-800.
[vii]liao,h.;kadjo,a.f.;dasgupta,p.k.anal.chem.2015,87,8342-8346.
[viii]liu,y.;srinivasan,k.;pohl,c.;avdalovic,n.j.biochem.biophys.methods2004,60,205-232.
[ix]strong,d.l.;dasgupta,p.k.anal.chem.1989,61,939-945.
[x]al-horr,r.;dasgupta,r.;adams,r.l.anal.chem.2001,73,4694-4703.
[xi]waiz,s.;cedillo,b.m.;jambunathan,s.;hohnholt,s.g.;dasgupta,p.k.;wolcott,d.w.anal.chim.acta2001,428,163-171.
[xii]small,h.ionchromatographyplenum,newyork,1989,pp.139-148.
[xiii]zini,q.;buldini,p.l.;morettini,l.microchem.j.1985,32,148-152.
[xiv]americansocietyfortestingandmaterials.standardspecificationofreagentwater.astmd1193-06(reapproved2011).www.astm.org/standards/d1193.htm.
[xv]li,y.;muo,y.;xie,h.anal.chim.acta2002,455,315-325.
[xvi]sabarudin,a.;oshima,m.;ishii,n.;motomizu,s.talanta2003,60,1277-1285.
[xvii]ma,j.;adornato,l.;byrne,r.h.;yuan,d.trac,trendsanal.chem.2014,60,1-15.
[xviii]amornthammarong,n.;zhang,j.talanta2009,79,621-626.
[xix]rimmelin-maury,p.;moutin,t.;queguiner,b.anal.chim.acta2007,587,281-286.
[xx]ma,j.;byrne,r.h.talanta2012,88,484-489.
[xxi]hioki,a.;lam,j.w.h.;mclaren,j.w.anal.chem.1997,69,21-24.
[xxii]li,h.;chen,f.j.chromatogr.a2000,874,143-147.
- 還沒有人留言評論。精彩留言會獲得點贊!