采用一測多評法測定大葉金花草成分的方法與流程
本發明涉及一種質量檢測方法,具體涉及一種采用一測多評法測定大葉金花草成分的方法。
背景技術:
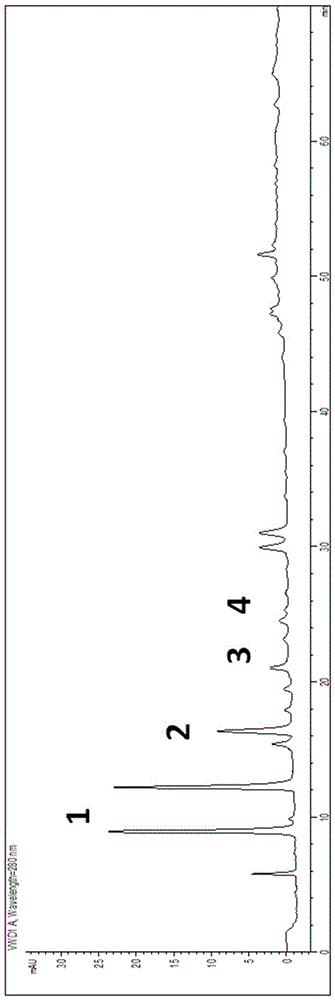
:大葉金花草出自《廣西中藥志》。大葉金花草多年生草本,高可達65厘米。根莖堅硬而短,橫走,密被赤褐色鉆狀鱗片。葉近生,葉柄長達25厘米,禾稈色,光亮,直立;葉近革質,無毛;3~4回羽狀分裂,披針形,長20~40厘米,寬5~12厘米;下部羽片卵狀技針形,斜展,長5~10厘米,寬2~5厘米;小羽片矩圓形或披針形;末回裂片楔形,先端截形,有牙齒,基部楔形,下延,葉脈下面明顯,2叉狀分枝。孢子囊群頂生,每裂片上1~2枚,囊群蓋灰棕色,半杯形,寬與葉緣等長,向外開裂。拉丁植物動物礦物名:Sphenomerischinensis(L.)Maxon[AdiantumchusanumL.;Stenolomachusana(L.)Ching]。拉丁文名:HerbaStenolomae。功效分類:清熱解毒藥;利濕藥。科屬分類:鱗始蕨科。別名:野黃連、水黃連、牙齒芒、擎天蕨、雪仙草、掃雪花、蜢蚱參、上樹細辛草、青蕨、金花草、大金花草、烏韭、石發、地柏枝、雉雞尾、小雞尾草、細葉狼箕、花葉鳳尾草、烏竹、墻柏、細葉鳳凰尾、土黃連、孔雀尾、萬能解毒草、苦黃連。性味:微苦;寒;無毒。歸經:肝經;肺經;大腸經。功能:清熱解毒;利濕止血。主治:感冒發熱;咳嗽;咽喉腫痛;腸炎;痢疾;肝炎;濕熱帶下;癰瘡腫毒;痄腮;口瘡;燙火傷;毒蛇、狂犬咬傷;皮膚濕疹;吐血;尿血;便血;外傷出血。用法用量:內服:煎湯,15-30g;或絞汁。外用:適量,搗敷;或研末外敷;或煎湯洗。生態環境:生于海拔200-1900m的林下、路邊或空曠處。藥材基源:為鱗始蕨科植物烏蕨的全草或根莖。采收儲藏:夏、秋季挖取帶根莖的全草,去雜質,洗凈,鮮用或曬干。大葉金花草廣泛的分布于西南及江蘇、安徽、浙江、江西、福建、臺灣、湖北、湖南、廣東、廣西、西藏、陜西、四川等地,具有清熱解毒、利濕、止血的功效、主治感冒發熱、咳嗽、咽喉腫痛、腸炎、痢疾、肝炎、濕熱帶下、癰瘡腫毒、痄腮、口瘡、燙火傷、毒傷、狂犬咬傷、皮膚濕疹、吐血、尿血、便血、外傷出血,具有廣譜的的抗菌活性和解毒作用。民間用于治療胃癌、腸癌、食物中毒和農藥中毒,有“萬能解毒藥”之稱。其化學成分主要有有機酸類和黃酮類。牡荊素又名牡荊苷,為大葉金花草的有效成分之一,具有防癌抗腫瘤、抗炎、解痙、降壓等作用。由于植物化學成分的復雜性,多指標含量測定已經成為植物來源藥物質量控制的共識。目前,越來越多的國內外植物藥標準將多指標含量測定收錄其中。然而,在多指標含量測定方法實施的過程中,對照品消耗量大,檢測操作難度高,嚴重制約了該方法的推廣應用。為解決以上問題,人們積極尋找替代性方法。研究人員發現,不同物質在紫外檢測器中的響應-濃度比的比值是一個常數,并將該常數定義為相對校正因子。在樣品檢測過程中,使用外標法測定A化合物的峰面積并獲得其含量,同時測定B化合物的峰面積,便可以根據以上信息和A/B物質間相對校正因子,計算獲得B化合物的含量。該替代對照品法被命名為一測多評法。由于大大減少了對照品的用量,并降低了檢測試驗的操作難度,一測多評質量控制方法得到迅速的推廣和應用。目前,美國藥典約有20余個品種,歐洲藥典約有10余個品種,中國藥典有1個品種(黃連),使用該方法對植物來源藥物進行多指標含量測定,且數目仍在不斷增加。原兒茶酸、原兒茶醛、葒草苷和牡荊素是大葉金花草中的主要成分,目前還沒有文獻公開將一測多評方法用于其中。技術實現要素:本發明的目的在于提供一種采用一測多評法測定大葉金花草成分的方法,針對現有單一成分和多指標含量測定方法的不足,本發明提供一種高實用性、操作簡單、節約成本的針對大葉金花草中原兒茶酸、原兒茶醛、葒草苷和牡荊素四種成分的檢測方法。為實現上述目的,本發明采用如下技術方案:采用一測多評法測定大葉金花草成分的方法,采用廉價易得的原兒茶酸作為內參物進行一測多評,建立原兒茶酸與大葉金花草中原兒茶酸、原兒茶醛、葒草苷和牡荊素之間的相對校正因子,通過校正因子計算大葉金花草中原兒茶酸、原兒茶醛、葒草苷和牡荊素的含量,采用液相色譜法進行測定,具體方法如下:(1)材料準備:供試品的大葉金花草全采自廣西,經廣西中醫藥大學藥用植物園鑒定為陵齒蕨科植物烏蕨StenolomachusanaumChing的全草;將供試品磨成粉末,過2號篩備用;(2)對照品溶液的制備:原兒茶醛:精密稱取,用50%乙醇溶解,制成濃度為0.0498mg/ml的對照溶液;原兒茶酸:五氧化二磷減壓干燥24h,精密稱定,用50%乙醇溶解,制成濃度為0.0538mg/ml的對照溶液;牡荊素:五氧化二磷減壓干燥24h,精密稱定,用50%乙醇溶解,制成濃度為0.0573mg/ml的對照溶液;葒草苷:五氧化二磷減壓干燥24h,精密稱定,用50%乙醇溶解,制成濃度為0.0503mg/ml的對照溶液;取上述對照品各1mL混合搖勻,作為混合對照品溶液;(3)供試品溶液的制備:稱取供試品粉末0.3g,置于50ml錐形瓶中,加入65%乙醇30ml超聲30min,濾過,揮干溶劑得殘留物,殘留物用10%乙腈定容至5ml容量瓶中,用移液管從容量瓶中移取1ml~5ml容量瓶中,再用10%乙腈稀釋至刻度線即得供試品溶液;(4)色譜條件及系統適用性試驗:色譜柱:DiamonsilC18;流動相:B泵:乙腈;D泵:0.4%磷酸;梯度洗脫程序:0~10min,B泵:10%→15%,D泵:90%→85%;10~40min,B泵:15%→20%,D泵:85%→80%;40~45min,B泵:20%→25%,D泵:80%→75%;45~55min,B泵:25%→30%,D泵:75%→70%;55~70min,B泵:30%→30%,D泵:70%→70%;檢測波長:210nm;流速:1.0mL/min;柱溫:25℃;理論塔板數按原兒茶酸計算不低于15000;(5)測定:將供試品溶液放于離心機中13000r/min離心10分鐘,精密吸取上清液10μL,注入液相色譜儀,采用內標法計算,供試品溶液的特征圖譜中呈現原兒茶酸、原兒茶醛、葒草苷和牡荊素的峰值,通過與原兒茶酸的相對校正因子計算原兒茶酸、原兒茶醛、葒草苷和牡荊素的含量。所述液相色譜儀為PE200系列高效液相色譜儀,選擇耐受壓力為0-6200psi或0-42MPa。取步驟(2)混合對照品溶液,分別進樣3、6、10、12、15μL測定,以原兒茶酸為內標,分別計算原兒茶醛、葒草苷、牡荊素的相對校正因子fR,,采用DiamonsilC18色譜柱分別考察Agilent1260、Waters2695-2998液相色譜系統對fR的影響,上述四個成分的相對標準偏差RSD<3%,從中確定原兒茶醛、葒草苷、牡荊素的相對校正因子fR為各實驗所得數據的平均值,分別是0.82、0.55、0.60。本發明的有益效果為:1、本發明選用原兒茶酸作為內參物進行QAMS(一測多評法),原兒茶酸價廉容易得到,從而可以節省時間成本、勞動成本,而且方法快速、準確,能更全面的知曉大葉金花草質量;2、針對現有單一成分和多指標含量測定方法的不足,本發明檢測方法高實用性、操作簡單、節約成本,可以有效檢測出大葉金花草中原兒茶酸、原兒茶醛、葒草苷和牡荊素四種成分的含量;3、通過比較一測多評法和外標法的結果差異,利用Spss20.0軟件對Pearson相關系數進行比較,兩種方法所得含量結果相近,計算兩種方法的相對誤差,結果絕對值<0.3%,說明一測多評所得的含量計算結果與外標法所得結果相似,在不同的實驗條件下,各成分之間的校正因子重現性好(RSD<3%),能夠有效進行大葉金花草中原兒茶酸、原兒茶醛、葒草苷和牡荊素四種成分的含量測定;4、在對照缺乏的情況下,本發明的方法可以應用于大葉金花草的質量評價,同時測定金花草中4個化合物的方法簡便、快捷,為建立金花草藥材的質量評價提供依據;5、構建了大葉金花草的一測多評檢測方法,由此可以更好更全面的控制大葉金花草的質量,同時規避由多成含量測定帶來的成本和操作問題,方法簡單便于操作,且測定結果準確,方法重復性,耐用性好。附圖說明圖1為采用水回流提取(乙酸乙酯萃取)對供試品進行HPLC分析得到的指紋圖譜;圖2為采用水超聲提取(乙酸乙酯萃取)對供試品進行HPLC分析得到的指紋圖譜;圖3為采用65%乙醇超聲提取(乙酸乙酯萃取)對供試品進行HPLC分析得到的指紋圖譜;圖4為采用65%乙醇超聲提取對供試品進行HPLC分析得到的指紋圖譜;圖5為采用乙酸乙酯超聲提取對供試品進行HPLC分析得到的指紋圖譜;圖6為采用水冷浸法提取對供試品進行HPLC分析得到的指紋圖譜;圖7為采用40%乙醇超聲提取(乙酸乙酯萃取)對供試品進行HPLC分析得到的指紋圖譜;圖8為原兒茶酸線性關系考察圖;圖9為原兒茶醛線性關系考察圖;圖10為葒草苷線性關系考察圖;圖11為牡荊素線性關系考察圖;圖12采用一測多評法與外標法測定結果比較金花草中四種物質含量測定結果(n=3)圖。具體實施方式采用一測多評法測定大葉金花草成分的方法,采用廉價易得的原兒茶酸作為內參物進行一測多評,建立原兒茶酸與大葉金花草中原兒茶酸、原兒茶醛、葒草苷和牡荊素之間的相對校正因子,通過校正因子計算大葉金花草中原兒茶酸、原兒茶醛、葒草苷和牡荊素的含量,采用液相色譜法進行測定,具體方法如下:(1)材料準備:供試品的大葉金花草全采自廣西,經廣西中醫藥大學藥用植物園鑒定為陵齒蕨科植物烏蕨StenolomachusanaumChing的全草;將供試品磨成粉末,過2號篩備用;(2)對照品溶液的制備:原兒茶醛:精密稱取,用50%乙醇溶解,制成濃度為0.0498mg/ml的對照溶液;原兒茶酸:五氧化二磷減壓干燥24h,精密稱定,用50%乙醇溶解,制成濃度為0.0538mg/ml的對照溶液;牡荊素:五氧化二磷減壓干燥24h,精密稱定,用50%乙醇溶解,制成濃度為0.0573mg/ml的對照溶液;葒草苷:五氧化二磷減壓干燥24h,精密稱定,用50%乙醇溶解,制成濃度為0.0503mg/ml的對照溶液;取上述對照品各1mL混合搖勻,作為混合對照品溶液;(3)供試品溶液的制備:稱取供試品粉末0.3g,置于50ml錐形瓶中,加入65%乙醇30ml超聲30min,濾過,揮干溶劑得殘留物,殘留物用10%乙腈定容至5ml容量瓶中,用移液管從容量瓶中移取1ml~5ml容量瓶中,再用10%乙腈稀釋至刻度線即得供試品溶液;(4)色譜條件及系統適用性試驗:用DiamonsilC18色譜柱;乙腈-磷酸鹽緩沖液為流動相按以下梯度運行條件洗脫,流速:1.0mL/min,柱溫:25℃,檢測波長:210nm;理論塔板數按原兒茶酸計算不低于15000,流動相條件見表1:表1大葉金花草高效液相流動相條件時間(min)乙腈/%0.4%磷酸(%)0~1010→1590→8510~4015→2085→7040~4520→2570→7545~5525→3075→7055~7030→3070→70(5)測定:將供試品溶液放于離心機中13000r/min離心10分鐘,精密吸取上清液10μL,注入液相色譜儀,采用內標法計算,供試品溶液的特征圖譜中呈現原兒茶酸、原兒茶醛、葒草苷和牡荊素的峰值,通過與原兒茶酸的相對校正因子計算原兒茶酸、原兒茶醛、葒草苷和牡荊素的含量。提取方法和提取溶劑的確定由于葒草苷和牡荊素屬于黃酮苷類,具有一定的水溶性,可溶于熱水、甲醇、乙醇、乙酸乙酯等;原兒茶酸溶于水、乙醇、乙醚;原兒茶醛易溶于乙醇、丙酮、乙酸乙酯、乙醚、熱水和冷水,不溶于苯和氯仿等溶劑,故可以用于提取以上物質的溶劑有:水、乙醇、乙酸乙酯。常用的溶劑提取方法有:煎煮、回流、超聲、冷浸等。本實驗對提取方法進行了如下比較:取04號大葉金花草藥材粉末0.3g(共7份),分別采用水回流提取,乙酸乙酯萃取;水超聲提取,乙酸乙酯萃取;65%乙醇超聲提取,乙酸乙酯萃取;65%乙醇超聲提取;乙酸乙酯超聲提取;水冷浸法提取;40%乙醇超聲提取,乙酸乙酯萃取(均為20ml溶劑,超聲和回流均為30min,冷浸為3h,萃取均為40ml乙酸乙酯,萃取兩次,每次20ml)。超聲功率為200W,頻率40kHz。過濾,將溶液濃縮至干,10%乙腈溶解,定容至5ml容量瓶中,用移液管從容量瓶中移取1ml至5ml容量瓶中,再用10%乙腈稀釋至刻度線。進高效液相儀檢測前,先用相應濾頭濾過,進行HPLC分析,:用DiamonsilC18色譜柱;乙腈-磷酸鹽緩沖液為流動相按以下梯度運行條件洗脫,流速:1.0mL/min,柱溫:25℃,檢測波長:210nm;理論塔板數按原兒茶酸計算不低于15000,流動相條件見表1。結果如下(圖1-7)。結果表明:方法牡荊素沒有出峰,方法基線不平,峰形不好,對照物質沒有完全出峰,方法、牡荊素峰太小,易受噪音干擾,方法、、對照物質皆出峰,基線較好,通過比較四種對照物質的峰面積,方法65%乙醇超聲提取的方法四種對照物質峰面積皆大于其它方法,基線平穩,故選擇該法對藥材進行提取。表2方法、、號4種物質面積最佳吸收波長的確定四種對照物質的掃描光譜顯示,在210nm附近皆有最大吸收,因此,檢測波長定為210nm。比較了0.5%、0.4%和0.3%的磷酸,三種濃度下四個對照品峰的分離度和對稱因子,綜合考慮兩因素,以及高濃度磷酸溶液對色譜柱的傷害,最后選擇0.4%的磷酸溶液為流動相,如表3。表3不同磷酸濃度分離效果柱溫考察比較了35℃、25℃和15℃三個柱溫下四個對照品峰的分離度和對稱因子,綜合考慮兩因素,最后選擇25℃未檢測溫度,如表4。表4不同柱溫下分離情況原兒茶酸線性關系考察取濃度為0.2150mg/mL的原兒茶酸對照,用50%乙醇分別稀釋成0.0215mg/mL、0.0043mg/mL、0.0022mg/mL和0.0011mg/mL的不同濃度原兒茶酸標準溶液,分別取10μL進樣分析。以對照品濃度(X)為橫坐標,峰面積(Y)為縱坐標,繪制標準曲線,進行線性回歸。結果顯示原兒茶酸標準曲線方程為Y=72003X+61.485,r2=0.9994,由此說明在0.0011mg/mL~0.2150mg/mL范圍內線性關系良好,見圖8。2.4.1.2原兒茶醛線性關系考察取濃度為0.1993mg/mL的原兒茶醛對照,用50%乙醇分別稀釋成0.0199mg/mL、0.0040mg/mL、0.0020mg/mL和0.0010mg/mL的不同濃度原兒茶醛標準溶液,分別取10μL進樣分析。以對照品濃度(X)為橫坐標,峰面積(Y)為縱坐標,繪制標準曲線,進行線性回歸。結果顯示原兒茶醛標準曲線方程為Y=56924X+27.845,r2=0.9996,由此說明在0.0010mg/mL~0.1993mg/mL范圍內線性關系良好,見圖9。2.4.1.3葒草苷線性關系考察取濃度為0.2290mg/mL的葒草苷對照,用50%乙醇分別稀釋成0.0229mg/mL、0.0046mg/mL、0.0023mg/mL和0.0011mg/mL的不同濃度葒草苷標準溶液,分別取10μL進樣分析。以對照品濃度(X)為橫坐標,峰面積(Y)為縱坐標,繪制標準曲線,進行線性回歸。結果顯示葒草苷標準曲線方程為Y=44958X-19.379,r2=1,由此說明在0.0011mg/mL~0.2290mg/mL范圍內線性關系良好,見圖10。2.4.1.4牡荊素線性關系考察取濃度為0.2013mg/mL的原兒茶醛對照,用50%乙醇分別稀釋成0.0201mg/mL、0.0040mg/mL、0.0020mg/mL和0.0010mg/mL的不同濃度牡荊素標準溶液,分別取10μL進樣分析。以對照品濃度(X)為橫坐標,峰面積(Y)為縱坐標,繪制標準曲線,進行線性回歸。結果顯示牡荊素標準曲線方程為Y=54075X+2.343,r2=1,由此說明在0.0010mg/mL~0.2013mg/mL范圍內線性關系良好,見圖11。穩定性試驗取04號樣品的供試品溶液,于制備后0h、3h、6h、9h和12h分別進樣10μL,分別測定原兒茶酸、原兒茶醛、葒草苷和牡荊素的峰面積,計算原兒茶酸RSD=0.99%、原兒茶醛RSD=1.59%、葒草苷RSD=1.61%、牡荊素RSD=1.17%,表明樣品在12h以內穩定。專屬性樣品在相應對照品的保留時間附近出峰,且分離度和對稱因子符合含量測定相關要求;空白進樣檢測表明溶劑對四種物質檢測沒有影響;延長沖洗至100min,在70min后沒有峰出現。加樣回收率試驗精密稱取已知含量的04號粉末(過20目,原兒茶酸、原兒茶醛、葒草苷、牡荊素含量分別為1.07mg/g、0.31mg/g、0.59mg/g、0.97mg/g)0.3g共6份,分別置50ml錐形瓶中,精密加入濃度為0.2150mg/ml的原兒茶酸對照品溶液2ml、濃度為0.1993mg/ml的原兒茶醛對照品0.5ml、濃度為0.2290mg/ml的葒草苷對照品1ml以及濃度為0.2013mg/ml的牡荊素對照品溶液0.5ml,按“2.1.2”項下方法制備所需溶液,按“2.2”項色譜條件,分別進樣10μL,測定樣品中四種物質的含量,計算平均加樣回收率(n=6)和RSD,結果見表5-。表5加樣回收率試驗數據表(原兒茶酸)表6加樣回收率試驗數據表(原兒茶醛)表7加樣回收率試驗數據表(葒草苷)表8加樣回收率試驗數據表(牡荊素)待測組分fR的計算取混合對照品溶液,分別進樣3、6、10、12、15μL測定,以原兒茶酸為內標,分別計算原兒茶醛、葒草苷、牡荊素的fR,結果見表9。表9四種成分的fR(n=3)fR的重現性考察采用Agilent1260分別考察3種不同色譜柱(PhenomenexGemini5μmC18、依利特SinoChromOBS-BP5μm和AgilentTC-C18)對校正因子重現性進行考察;采用DiamonsilC18色譜柱分別考察了Agilent1260、Waters2695-2998液相色譜系統對fR的影響,結果4個成分的校正因子重現性良好(RSD<3%)。結果見表10。表10fR重現性考察(n=3)fR的確定依據《一測多評法建立的技術指南》和上述實驗結果,最終確定原兒茶醛、葒草苷、牡荊素的fR為各實驗所得數據的平均值,分別是0.82、0.55、0.60。待測成分色譜峰的定位計算上述不同色譜儀器和不同色譜柱中各待測成分與原兒茶酸的相對保留時間,對其余3個待測成分進行定位。計算結果表明相對保留時間的波動較小(RSD<1%),結果見表11。表11各成分的相對保留時間(n=3)一測多評法與外標法測定結果比較對25批廣西產金花草按照前述方法制備供試品溶液,每個產地樣品制備三份,按前述色譜條件進行測定,以一測多評法和外標法計算樣品的含量(參見圖12)。結果顯示廣西各地產金花草藥材全草中原兒茶酸、原兒茶醛、葒草苷和牡荊素的范圍分別為0.43~1.50mg/g、0.20~0.68mg/g、0.37~0.92mg/g和0.12~0.97mg/g;葉中四個化合物含量范圍分別為0.31~1.15mg/g、0.17~0.52mg/g、0.27~0.97mg/g和0.18~0.54mg/g;金花草藥材根及根莖中四個化合物含量范圍分別為0.17~0.59mg/g、0.04~0.14mg/g、0.28~0.67mg/g和0.08~0.10mg/g;此結果顯示地下部分四種物質含量范圍低于地上部分。結論原兒茶酸、原兒茶醛、葒草苷和牡荊素是大葉金花草中的主要成分,有研究分別指出了其的藥理作用,因此選擇它們作為大葉金花草藥材質量評價的指標成分。本實驗選用原兒茶酸作為內參物進行QAMS(一測多評法)是由于原兒茶酸作為水(醇)提物,是大葉金花草中主要的有效成分,在大葉金花草中含量較高,且該對照品價廉容易得到。為比較一測多評法和外標法的結果差異,利用Spss20.0軟件對Pearson相關系數進行比較,原兒茶醛、葒草苷和牡荊素的相關系數分別為0.997、0.998和0.999,可見兩種方法所得含量結果相近,計算兩種方法的相對誤差,結果絕對值<0.3%,說明一測多評所得的含量計算結果與外標法所得結果相似,在不同的實驗條件下,各成分之間的校正因子重現性好(RSD<3%),說明在對照缺乏的情況下,一測多評法可以應用于大葉金花草的質量評價,同時測定金花草中4個化合物的方法簡便、快捷,本實驗結果為建立金花草藥材的質量評價提供依據。對于不同采收時間:比較第08號和09號樣品,對于同一個產地的大葉金花草,冬季采收四種成分含量是春季的約2倍見表12。對于不同用藥部位:對于同一批藥材,比較10號和11號,12號和13號,14號和15號,對原兒茶醛和牡荊素根及根莖的含量更高,而原兒茶酸和葒草苷地上部分含量高于根及根莖,見表13。表12大葉金花草不同采收時間含量測定結果表13大葉金花草不同部位含量測定結果當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1