一種奈帕芬胺有關物質的RT?HPLC檢測方法與流程
本發明涉及藥物分析技術領域,特別涉及一種奈帕芬胺及其降解雜質和工藝雜質的反向高效液相色譜檢測方法。
背景技術:
奈帕芬胺,化學名為2-氨基-3-苯甲酰基苯基乙酰胺,其結構式如下:
奈帕芬胺(Nepafenac)是一種新型眼用非甾體類解熱鎮痛抗炎藥(NSAIDs)。用于眼內炎癥的治療。目前研究結果表明Nepafenac效果優于傳統NSAIDs且安全性良好。2005年8月19日,美國食品與藥物管理局(FDA)允許其眼用混懸液(商品名為Nevanac,Alton公司)應用于臨床,使其成為FDA批準的首個眼科用非激素類抗炎前體藥物。
Nepafenac經眼部給藥后,可迅速穿過角膜,并在眼組織水解酶的作用下轉化為氨芬酸,迅速到達靶位點發揮作用。而氨芬酸又通過抑制前列腺素H合成酶(環氧化酶),阻斷前列腺素的合成,來發揮其抗炎止痛的作用。NSAIDs經眼部給藥后,可抑制前列腺素在虹膜、睫狀體和結膜的合成,因此可預防眼部炎癥的發生,減少相關的疼痛。與傳統NSAIDs相比,Nepafenac具有滲透力強、靶向作用強、毒副作用小等優點。
在藥物分析中所稱的“有關物質(related substances)”是指在原料藥生產過程中帶入的起始原料、試劑、中間體、副產物和異構體等物質,也可能是制劑在生產、貯藏和運輸過程中產生的降解產物、聚合物或晶型轉變等特殊雜質。有關物質的種類與藥物的合成路線和制劑工藝密切相關,藥物在合成和制劑過程中的任何一個因素的改變都可能導致其有關物質的種類不同,因而有關物質的檢測和控制過程相對復雜。有關物質的檢測是控制藥品質量的重要指標。
在奈帕芬胺原料藥生產過程中帶入的起始原料、試劑、中間體、副產物和異構體等雜質,確定相關雜質譜:雜質A、B、C、D、E、F、G、H、I(見表1)。
表1奈帕芬胺各雜質名稱及結構
現有文獻中為見能夠分離上述8種雜質的分析方法,本發明提供一種能夠分離上述雜質和奈帕芬胺的分析方法。
技術實現要素:
本發明提供一種高靈敏度的檢測方法,解決將奈帕芬胺特征峰與降解雜質、較多的合成中間體及工藝雜質特征峰分離的問題。
本發明技術方案如下:
奈帕芬胺的有關物質雜質的RT-HPLC檢測方法,包括下列步驟:
a)配置分析溶液
用甲醇、乙腈單一溶劑或甲醇-水、乙腈-水混合溶液溶解樣品,制成分析溶液;
b)色譜條件
色譜柱為反相色譜柱,所述反相色譜柱選自苯基硅烷鍵合硅膠色譜柱、十八烷基硅烷鍵合硅膠色譜柱或八烷基硅烷鍵合硅膠色譜柱;
以酸銨鹽溶液-有機相的混合液為流動相,流動相pH范圍5~8,所述的有機相為甲醇或乙腈;采用梯度洗脫方法,流速為0.8-1.3ml/min;柱溫為25-50℃,檢測波長為205-300nm;運行時間40-70min;
c)上機測定
取步驟a)制成的分析溶液5-40μl注入高效液相色譜儀,進行色譜分析,并記錄色譜圖。
在一些實施例中,所述反相色譜柱的填充劑為苯基硅烷鍵合硅膠色譜柱或十八烷基硅烷鍵合硅膠色譜柱或八烷基硅烷鍵合硅膠色譜柱,優選為十八烷基硅烷鍵合硅膠色譜柱。
在一些實施例中,所述反相色譜柱的規格為:柱長介于100mm至300mm,色譜柱內徑介于1mm至10mm,粒徑介于1μm至10μm,優選為Agilent ZORBAX C18(4.6mm×250mm,5μm)的十八烷基硅烷鍵合硅膠色譜柱。
在一些實施例中,所述酸銨鹽溶液的濃度為0.001-0.05mol/L、pH 2.0-7.0,優選酸銨鹽溶液的濃度為0.01mol/L、pH 6.5。
在一些實施例中,所述酸銨鹽為甲酸銨、乙酸銨、碳酸氫銨中之一,優選乙酸銨。
所述酸銨鹽溶液-有機相的體積比為90:10-20:80。在一些實施例中,所述酸銨鹽溶液-有機相,所述酸銨鹽溶液-有機相的體積比為90:10~30:70,優選為70:30~30:70。
所述步驟b為所述反相色譜柱為十八烷基硅烷鍵合硅膠色譜柱,所述流動相為乙酸銨溶液與乙腈混合液,乙酸銨溶液0.01mol/L,所述流動相中乙酸銨溶液-乙腈的比例按0、25~30、38~42、48~52、53~57、63~67min時間點,水相體積比為73~77%、73~77%、58~62%、13~16%、13~16%、73~77%、進行梯度洗脫。。
所述樣品為奈帕芬胺原料藥或其制劑,所述制劑為片劑、膠囊劑、顆粒劑、眼用制劑、鼻用制劑、栓劑、丸劑、軟膏劑乳膏劑、糊劑、吸入制劑、噴霧劑、氣霧劑、凝膠劑、散劑、糖漿劑、搽劑、涂劑、涂膜劑、酊劑、貼劑、口服溶液劑、植入劑、膜劑、洗劑、沖洗劑、煎膏劑、膏藥、露劑、茶劑。
有益效果:
本發明能夠有效的測定奈帕芬胺溶液中各種相關雜質的存在,尤其實現了譜圖中奈帕芬胺特征峰與奈帕芬胺降解雜質特征峰及其相關中間體、工藝雜質特征峰的分離。同時本發明的方法還可用于奈帕芬胺制劑的有關物質檢測。
本發明的分析方法各雜質的分離度都大于1.5,不對稱因子都在1.9-1.2范圍內,達到準確有效分離目的。上述方法的檢測可以確保產品的質量可控。
附圖說明
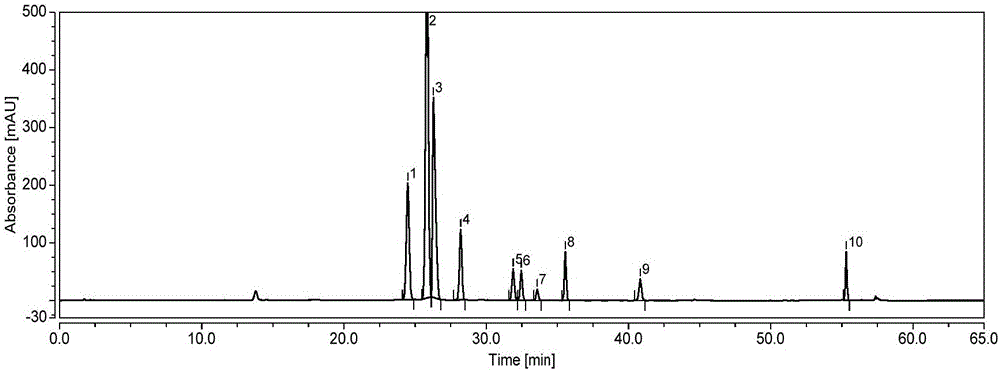
圖1實施例1方法梯度洗脫的色譜圖;
(注:圖中1‐10分別為雜質A、B、C、奈帕芬胺、D、E、F、G、H、I)
圖2實施例1方法梯度洗脫的色譜圖;
(注:圖中1‐10分別為雜質A、B、C、奈帕芬胺、D、E、F、G、H、I)
圖3實施例1方法梯度洗滌的色譜圖;
(注:圖中1‐10分別為雜質A、B、C、奈帕芬胺、D、E、F、G、H、I)
圖4實施例1方法梯度洗滌的色譜圖;
(注:圖中1‐10分別為雜質A、B、C、奈帕芬胺、D、E、F、G、H、I)。
具體實施方式
以下結合具體實施例,進一步闡述本發明。這些實施例僅用于說明本發明而不用于限制本發明的范圍。下列實施例中未注明具體條件的試驗方法,通常按照常規條件或按照制造廠商所建議的條件。除非另行定義,文中所使用的所有專業與科學用語與本領域技術人員所熟知的意義相同。此外,任何與所記載內容相似或均等的方法及材料皆可用于本發明方法中。文中所述的較佳實施方法與材料僅作示范之用。
實施例1
(1)儀器與色譜條件
高效液相色譜儀:u3000高效液相色譜系統及工作站;
色譜柱:Thermo Syncronis C18(4.6mm×250mm,5μm)的十八烷基硅烷鍵合硅膠色譜柱
配置0.01mol/L乙酸銨溶液、稀氨水調節pH至3.5為水相,流動相中水相-乙腈的比例按0、26、35、40、47、50、60min時間點,水相體積比為72%、72%、55%、15%、75%、75%,設置流速為1.0ml/min,檢測波長為240nm,柱溫50℃。
(2)實驗步驟
分別取奈帕芬胺、雜質A、B、C、D、E、F、G、H、I適量,用乙腈-乙酸銨水溶液(80:20)溶解并稀釋成每1ml中約含奈帕芬胺及各雜質10μg的混合溶液,作為分析溶液;
取上述分析溶液5μl,注入液相色譜儀,記錄色譜圖。結果見附圖1,雜質和奈帕芬胺基本可以分離開,但該條件下雜質B峰形較差,不對稱性因子較大0.7,一般而言不對稱性因子在0.9-1.2范圍內較為理想。雜質E、F、G未能實現基線完全分離。綜上,該方法需進行調整優化。
實施例2
(1)儀器與色譜條件
高效液相色譜儀:u3000高效液相色譜系統及工作站;
色譜柱:Thermo Syncronis C18(4.6mm×250mm,5μm)的十八烷基硅烷鍵合硅膠色譜柱;
配置0.01mol/L乙酸銨溶液、稀氨水調節pH至6.8為水相,流動相中水相-乙腈的比例按0、25、50、53、65min時間點,水相體積比為78%、78%、20%、78%、78%進行梯度洗脫,設置流速為1.3ml/min,檢測波長為240nm,柱溫40℃。
(2)實驗步驟
分別取奈帕芬胺、雜質A、B、C、D、E、F、G、H、I適量,用乙腈-水(70:30)
溶解并稀釋成每1ml中約含奈帕芬胺及雜質各20μg的混合溶液,作為分析溶液;
取上述分析溶液10μl,注入液相色譜儀,記錄色譜圖。結果見附圖2,可以看出該條件下奈帕芬胺主峰和其他雜質峰完全分離。
實施例3
(1)儀器與色譜條件
高效液相色譜儀:U3000高效液相色譜系統及工作站;
色譜柱:Agilent ZORBAX C18(4.6mm×250mm,5μm)的十八烷基硅烷鍵合硅膠色譜柱;
配置0.01mol/L乙酸銨溶液為水相,流動相中水相-乙腈的比例按0、28、50、52、60min時間點,水相體積比為75%、75%、25%、75%、75%進行梯度洗脫,流速1.1ml/min;柱溫35℃,檢測波長250nm;
(2)實驗步驟
分別取奈帕芬胺、雜質A、B、C、D、E、F、G、H、I適量,用乙腈-水(70:30)溶解并稀釋成每1ml中約含奈帕芬胺及雜質各10μg的混合溶液,作為分析溶液;
取上述分析溶液10μl,注入液相色譜儀,記錄色譜圖。結果見附圖3,可以看出該條件下萘帕芬胺主峰和其他雜質峰完全分離。
實施例4
(1)儀器與色譜條件
高效液相色譜儀:U3000高效液相色譜系統及工作站
色譜柱:Phenomenex Luna C8(4.6mm×250mm,5μm)的十八烷基硅烷鍵合硅膠色譜柱
配置甲醇-0.01mol/L乙酸銨溶液pH至6.0(10:90)為水相,流動相中水相-乙腈的比例按0、25、50、53、65min時間點,水相體積比為80%、80%、60%、20%、80%、80%,設置流速為1.0ml/min,檢測波長為240nm,柱溫50℃。
(2)實驗步驟
分別取奈帕芬胺、雜質A、B、C、D、E、F、G、H、I適量,用甲醇-水(70:30)溶解并稀釋成每1ml中約含奈帕芬胺各20μg的混合溶液,作為分析溶液;
取上述分析溶液30μl,注入液相色譜儀,記錄色譜圖。結果見附圖3,可以看出該條件下奈帕芬胺主峰和其他雜質峰完全分離。
如下表2所示,實施例1的方法分離出來結果,雜質B、F不對稱性較差,并且雜質E、F分離度為1.4和1.3,未完全分離。實施例2、3、4分離度和不對稱因子都比較好,能夠到達有效分離。
表2實施例1-4雜質分離度和不對稱因子情況表
- 還沒有人留言評論。精彩留言會獲得點贊!