氣相色譜?質譜法測定包裝材料中揮發性有機化合物的方法與流程
本發明屬于包裝材料檢測技術領域,特別是一種氣相色譜-質譜法測定包裝材料中揮發性有機化合物的方法。
背景技術:
目前,由于包裝材料在生產過程中使用大量的粘合劑、油墨和有機溶劑,這些輔助材料雖未與產品直接接觸,但在產品包裝和儲存過程中食品包裝材料中的VOCs(揮發性有機化合物)可能會遷移到產品和環境中,從而危害人體健康。而且,目前常見的包裝材料以高分子材料為主,高分子裂解產生的某些物質也可能會對人體及環境產生影響。因此,對包裝材料中VOCS的檢測控制正受到重視。目前,主要采用頂空采樣或固相微萃取采樣氣相色譜法分析。氣相色譜法分析法存在諸多不足:首先,固相微萃取采樣方式會引入有機溶劑,產生外來干擾;其次,采用氣相色譜法測定結果定性難度大,存在假陽性風險;最后,目前國內對包裝材料中溶劑殘留已有一些研究,但由于該方法測試種類少,已不能滿足目前對包裝材料中VOCs的測試要求。
技術實現要素:
本發明的目的在于克服現有技術的不足,提供一種氣相色譜-質譜法測定包裝材料中揮發性有機化合物的方法,有效減少外來干擾,提高對VOCs中各物質的定性能力。
本發明解決其技術問題是通過以下技術方案實現的:
一種氣相色譜-質譜法測定包裝材料中揮發性有機化合物的方法,其特征在于:所述方法的步驟為:
1)試樣制備:將待測樣品迅速裁成碎片置于頂空瓶中,密封后待測;
2)頂空進樣:將待測樣品送入頂空進樣器加熱套中加熱,頂空條件:加熱箱90-120℃,定量環90-120℃,傳輸線100-110℃,平衡時間25-40min;
3)色譜-質譜聯合測定:
用頂空進樣器將待測樣品注入氣相色譜-質譜聯用儀進行色譜-質譜聯合測定;
4)定性確證:
試樣待測液和已知標準物質的色譜峰在相同保留時間處出現,此時可定性確證目標分析物;
5)定量分析:
以各標準物質溶液體積為橫坐標,各自的定量色譜響應值為縱坐標,做標準曲線線性回歸方程,以試樣的響應值與標準曲線比較定量;
6)計算結果
溶劑殘留量按式(1)進行計算:
式中:
W……溶劑殘留量,單位為毫克每平方米(mg/m2)
P……對應量,單位為毫克(mg)
S……試樣面積、質量或體積,單位為平方米(m2)。
所述步驟3)中的色譜-質譜聯合測定的色譜條件為:HP-VOC(60m×0.32mm×1.80μm)毛細管色譜柱或同等色譜柱;載氣為高純氦氣;不分流進樣;氣體流速為2mL/min;柱溫35℃,保持1min;10℃/min升至180℃,保持5min;汽化室溫度250℃;接口溫度250℃;溶劑延遲3.5min;質譜條件:離子源溫度230℃;四極桿溫度150℃;電離方式:EI;電離能量:70eV;質量掃描范圍50~450amu;測定方式為SIM掃描方式;標準譜圖調諧。
本發明的優點和有益效果為:
1、本氣相色譜-質譜法測定包裝材料中揮發性有機化合物的方法,采用頂空采樣氣相色譜-質譜法進行定包裝材料中VOCs,采用頂空采樣方式,不用有機溶劑萃取和濃縮,減少了外來溶劑的干擾;使用氣相色譜-質譜聯用儀進行檢測,增強了方法定性能力,降低假陽性風險;能夠測試較多種類,包含醇類、酯類、酮類、苯類、鹵代烴等,基本滿足目前對包裝材料中VOCs的測試要求。
附圖說明
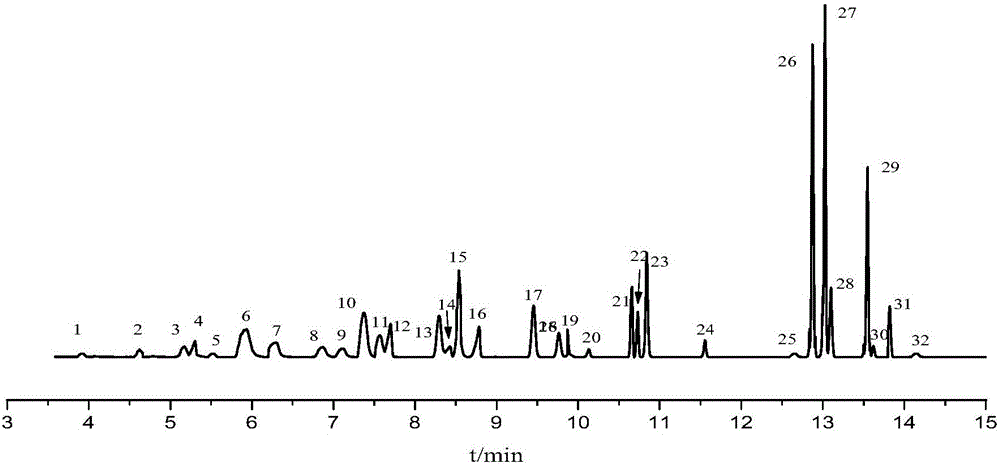
圖1為32種VOCs總離子流圖;
圖2為測試樣品VOCs SIM圖。
附圖標記說明
1甲醇;2乙醇;3丙酮;4異丙醇;51,1-二氯乙烯;6二氯甲烷;7丙醇;8正己烷;9丁酮;10乙酸乙酯;11三氯甲烷;12異丁醇;13乙酸異丙酯;14丁醇;15苯;16丙二醇甲醚;17乙酸正丙酯;18甲基環己烷;19環氧氯丙烷;20異戊醇;214-甲基-2-戊酮;22乙酸異丁酯;23甲苯;24乙酸正丁酯;25丙二醇甲醚醋酸酯;26乙苯;27間二甲苯;28對二甲苯;29苯乙烯;30鄰二甲苯;31環己酮;32異丙苯。
具體實施方式
下面通過具體實施例對本發明作進一步詳述,以下實施例只是描述性的,不是限定性的,不能以此限定本發明的保護范圍。
一種氣相色譜-質譜法測定包裝材料中揮發性有機化合物的方法,其方法的步驟為:
1)試樣制備:樣品:食品包裝復合膜;規格型號:材質:PA15/SPE60,尺寸(mm):150×85×0.075;樣品描述:來樣密封包裝,塑料卷材,樣品表面有印刷;測試內容:測試食品包裝材料VOCs。裁取材料中印刷圖案密集區域的100cm2樣品,將待測樣品迅速裁成碎片置于頂空瓶中,密封后待測。
2)頂空進樣:將待測樣品送入頂空進樣器加熱套中加熱,頂空條件:加熱箱100℃,定量環100℃,傳輸線105℃,平衡時間30min。
3)色譜-質譜聯合測定:
用頂空進樣器將待測樣品注入氣相色譜-質譜聯用儀進行色譜-質譜聯合測定;色譜條件為:HP-VOC(60m×0.32mm×1.80μm)毛細管色譜柱或同等色譜柱;載氣為高純氦氣;不分流進樣;氣體流速為2mL/min;柱溫35℃,保持1min;10℃/min升至180℃,保持5min;汽化室溫度250℃;接口溫度250℃;溶劑延遲3.5min。
質譜條件:離子源溫度230℃;四極桿溫度150℃;電離方式:EI;電離能量:70eV;質量掃描范圍50~450amu;測定方式為SIM掃描方式;標準譜圖調諧。
4)定性確證:
試樣待測液和已知標準物質的色譜峰在相同保留時間處出現,此時可定性確證目標分析物;
VOCs組分定量離子表
5)定量分析:
以各標準物質溶液體積為橫坐標,各自的定量色譜響應值為縱坐標,做標準曲線線性回歸方程,以試樣的響應值與標準曲線比較定量。
通過計算出測試樣品的響應值(見6中圖2);
圖中出現兩個峰,分別為異丁醇(1)和異戊醇(2),通過軟件計算響應值分別為138.57和990.71。
6)計算結果
溶劑殘留量按式(1)進行計算:
式中:
W……溶劑殘留量,單位為毫克每平方米(mg/m2)
P……對應量,單位為毫克(mg)
S……試樣面積、質量或體積,單位為平方米(m2)。
按照標準曲線計算得:0.45mg/m2和3.61mg/m2。
盡管為說明目的公開的本發明的實施例和附圖,但是本領域的技術人員可以理解,在不脫離本發明及所附權利要求的精神和范圍內,各種替換、變化和修改都是可能的,因此本發明的范圍不局限于實施例和附圖所公開的內容。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 用氣相色譜?正化學源?質譜聯用技術測定食用植物油中生育酚和生育三烯酚含量的方法與流程
- 一種超臨界流體色譜?氣相色譜?質譜測定卷煙主流煙氣中苯并[a]芘的方法與流程
- 一種超臨界流體色譜?氣相色譜?質譜測定卷煙主流煙氣中酚類化合物的方法與流程
- 一種用于氣相催化裂解制備偏二氯乙烯的催化劑的方法與流程
- 一種氣相色譜質譜聯用檢測水域中四溴雙酚A的方法與流程
- 用于二惡英在線檢測的氣相色譜?質譜間傳輸線系統的制造方法與工藝
- 一種氣相色譜?質譜法快速測定棉花纖維中4種有機氯農藥殘留的檢測方法與流程
- 一種用于二惡英在線檢測的氣相色譜?質譜間傳輸線系統的制造方法與工藝
- 一種檢測紡織品中聚丙烯酸及其酯類聚合物的分析方法與流程
- 血液中未知毒物的氣相色譜?質譜篩查方法與流程