一種替格瑞洛手性異構體含量的高效液相色譜檢測方法與流程
本發明屬于藥物化學中的分析監測
技術領域:
,更具體地講,涉及一種替格瑞洛手性異構體含量的高效液相色譜檢測方法。
背景技術:
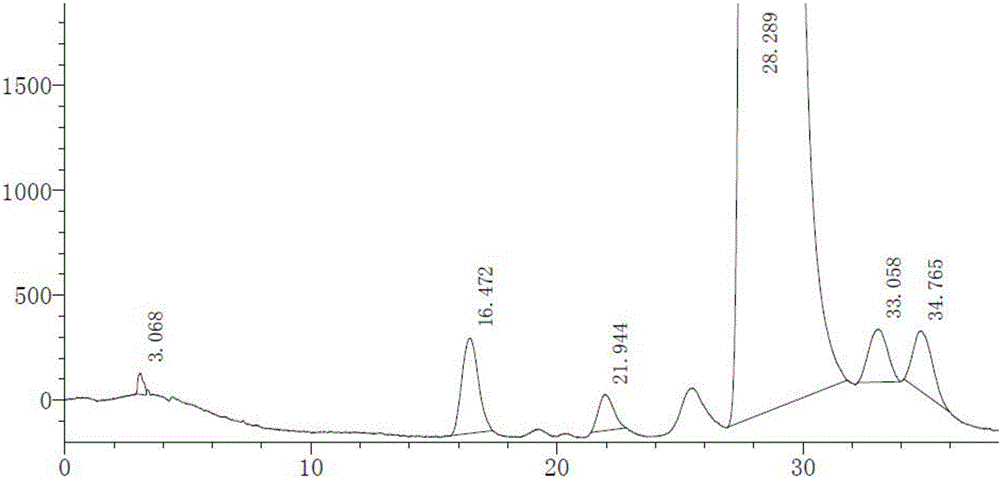
:替格瑞洛(ticagrelor)是由阿斯利康公司(AstraZeneca)研發的一種新型的、具有選擇性的小分子抗凝血藥,其化學結構式如下式1所示:替格瑞洛分子結構中含有6個手性碳原子,在實際合成過程中和儲藏過程中會產生雜質,其中手性異構體雜質是其主要雜質之一,而常規的分析檢測方法很難分離眾多的手性異構體雜質。已有文獻或專利報道采用纖維素類(或多糖衍生物)為填充劑的手性色譜柱檢測的方法,但均為硅膠表面涂敷纖維素類(或多糖衍生物)的柱子,此類柱子平衡時間較長、穩定性不好、重現性較差、柱子壽命較短,且無法使用二氯甲烷、三氯甲烷等溶劑為流動相。技術實現要素:為了解決現有技術中存在的問題,本發明的目的是提供一種能夠有效檢測替格瑞洛中手性異構體含量并保證藥物安全性和質量可靠性的替格瑞洛手性異構體含量的高效液相色譜檢測方法。本發明公開了一種替格瑞洛手性異構體含量的高效液相色譜檢測方法,采用以硅膠表面鍵合纖維素-三(3,5-二氯苯氨基甲酸酯)為填充劑的手性色譜柱,以低級烷烴-低級醇的混合溶液為流動相A,以二氯甲烷和/或氯仿為流動相B。根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,所述檢測方法具體包括以下步驟:a)分別稱取替格瑞洛對照品和替格瑞洛手性異構體對照品,利用無水乙醇溶解并稀釋制成每1ml中約含替格瑞洛1mg、各手性異構體2μg的系統適用性溶液;b)稱取替格瑞洛樣品,利用無水乙醇配制成每1ml中含1mg替格瑞洛的供試品溶液;c)利用無水乙醇將所述供試品溶液稀釋1000倍作為對照溶液;d)分別取步驟a)、步驟b)、步驟c)的溶液各10μl,分別注入高效液相色譜儀并進行測定,記錄色譜圖,完成替格瑞洛手性異構體的測定,采用主成份自身對照法對替格瑞洛手性異構體的含量進行計算;根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,流動相的流速為0.8~1.2ml/min,色譜柱柱溫為22~28℃;檢測器為紫外檢測器,檢測波長為250~350nm。根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,所述低級烷烴為正己烷、正戊烷、環己烷和正庚烷中的至少一種,所述低級醇為異丙醇、甲醇和乙醇中的至少一種,所述低級烷烴-低級醇的混合溶液中低級烷烴與低級醇的體積比為100:10~120:10。根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,所述流動相A為正己烷-異丙醇-甲醇的混合溶液,其中,正己烷-異丙醇-甲醇的體積比為(300~330):(17~12):(17~12)。根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,所述手性色譜柱為IC、IA和IB手性色譜柱中的一種。根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,按照下表所示的梯度進行洗脫:根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,替格瑞洛的手性異構體為下表所列的七種結構所示物質:根據本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的一個實施例,流動相的流速為1ml/min,檢測波長為300nm,手性色譜柱為IC,色譜柱柱溫為22℃、25℃或28℃。本發明采用高效液相色譜法并利用手性色譜柱對替格瑞洛手性異構體進行分離,式1中替格瑞洛的化合物色譜峰能有效的和其七種手性異構體實現分離,而且采用自身對照法進行雜質的定量測定,操作簡便、檢測成本低且具有良好的靈敏度、線性、專屬性、精密度、準確度、穩定性和耐用性,是控制替格瑞洛異構體的一種行之有效的檢測方法,從而保證了替格瑞洛產品質量和患者用藥安全。附圖說明圖1示出了實施例1中采用本發明的檢測方法對系統適用性溶液檢測得到的HPLC圖。圖2示出了實施例1中采用本發明的檢測方法對供試品溶液檢測得到的HPLC圖。圖3示出了對比例1中檢測方法對系統適用性溶液檢測得到的HPLC圖。圖4示出了對比例2中檢測方法對系統適用性溶液檢測得到的HPLC圖。圖5示出了對比例3中檢測方法對系統適用性溶液檢測得到的HPLC圖。圖6示出了對比例4中檢測方法對系統適用性溶液檢測得到的HPLC圖。具體實施方式本說明書中公開的所有特征,或公開的所有方法或過程中的步驟,除了互相排斥的特征和/或步驟以外,均可以以任何方式組合。本說明書中公開的任一特征,除非特別敘述,均可被其他等效或具有類似目的的替代特征加以替換。即,除非特別敘述,每個特征只是一系列等效或類似特征中的一個例子而已。下面將對本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法進行詳細地說明。根據本發明的示例性實施例,本發明采用以硅膠表面鍵合纖維素-三(3,5-二氯苯氨基甲酸酯)為填充劑的手性色譜柱,以低級烷烴-低級醇的混合溶液為流動相A,以二氯甲烷和/或氯仿為流動相B。本發明通過采用更穩定的硅膠表面鍵合纖維素-三(3,5-二氯苯氨基甲酸酯)為填充劑的手性色譜柱,解決硅膠表明涂敷纖維素類(或多糖衍生物)色譜柱壽命短,容易被試劑損傷(試劑的等級要求較高),耐用性較差的問題。雖然涂敷型色譜柱和鍵合型色譜柱僅僅是其固定相的固定方式存在差異,但是其對化合物的選擇性存在明顯的差異,因而屬于完全不同的色譜柱。替格瑞洛分子結構中含有6個手性碳原子,在實際合成過程中和儲藏過程中會產生雜質,其中手性異構體雜質是其主要雜質之一,而常規的分析檢測方法很難分離眾多的手性異構體雜質。而手性分離色譜柱眾多,主要是纖維素類、蛋白類,多糖,冠醚類。纖維素色譜柱主要分為涂敷型和鍵合型色譜柱,而目前替格瑞羅手性方法主要為涂敷型色譜柱,此類色譜柱溶劑兼容性窄,對溶劑以及稀釋劑有較大的限制;對流動相等級要求較高,部分國內廠家試劑無法滿足分析要求;壽命較短,從而造成色譜柱更換的耐用性較差。鍵合型色譜柱相對于涂敷型色譜柱,新增加的溶劑種類,給鍵合相特殊的選擇能力。因此本公司開發了鍵合相替格瑞羅手性開發方法,擴充了溶劑種類,可以兼容二氯甲烷和/或氯仿等大多數有機溶劑,因此不僅提高化合物的溶解性,同時也增加了對化合物獨特的選擇性,所以能夠有效分離替格瑞洛眾多的異構體雜質。而鍵合相色譜柱固有的穩定性和和耐用性,更增加了檢測方法的穩定性和重現性,從而使本發明遠好于目前采用涂敷型色譜柱方法。其中,替格瑞洛的化學結構式如下式1所示:并且,替格瑞洛的手性異構體為下表1所列的七種結構所示物質:表1替格瑞洛的七種手性異構體結構低級烷烴可以為正己烷、正戊烷、環己烷和正庚烷中的至少一種,低級醇可以為異丙醇、甲醇和乙醇中的至少一種,低級烷烴-低級醇的混合溶液中低級烷烴與低級醇的體積比為100:10~120:10。根據本發明的優選實施例,采用的流動相A為正己烷-異丙醇-甲醇的混合溶液,其中,正己烷-異丙醇-甲醇的體積比為(300~330):(17~12):(17~12)。本發明采用硅膠表面鍵合纖維素-三(3,5-二氯苯氨基甲酸酯)為填充劑的手性色譜柱(如:IC、IA和IB手性色譜柱中的任何一種)。具體地,本發明的檢測方法可以具體包括以下步驟:a)分別稱取替格瑞洛對照品和替格瑞洛手性異構體對照品,利用無水乙醇溶解并稀釋制成每1ml中約含替格瑞洛1mg、各手性異構體2μg的系統適用性溶液;b)稱取替格瑞洛樣品,利用無水乙醇配制成每1ml中含1mg替格瑞洛的供試品溶液;c)利用無水乙醇將所述供試品溶液稀釋1000倍作為對照溶液;d)分別取步驟a)、步驟b)、步驟c)的溶液各10μl,分別注入高效液相色譜儀并進行測定,記錄色譜圖,完成替格瑞洛手性異構體的測定,采用主成份自身對照法對替格瑞洛手性異構體的含量進行計算;其中,高效液相色譜的檢測條件具體為:流動相的流速為0.8~1.2ml/min,色譜柱柱溫為22~28℃;檢測器為紫外檢測器,檢測波長為250~350nm。進一步優選地,流動相的流速為1ml/min,檢測波長為300nm,手性色譜柱為IC,色譜柱柱溫為25℃。洗脫時,優選地采用下表2所示的梯度進行洗脫。表2洗脫程序根據現有要替格瑞洛生產路線和化合物的穩定性特點,主要控制7種手性異構體雜質,本發明采用高效液相色譜法并結合特定手性色譜柱進行分離,式1中替格瑞洛的化合物色譜峰能有效地和其7種手性異構體實現分離,而且,采用自身對照法進行雜質定量測定,操作簡便、檢測成本低且具有良好的靈敏度和專屬性,是輔助控制替格瑞洛手性異構體含量的有效檢測方法,從而保證了替格瑞洛的產品質量和患者用藥安全。下面將結合具體實施例和試驗例對本發明替格瑞洛手性異構體含量的高效液相色譜檢測方法的制備方法作進一步說明。實施例1:檢測儀器:島津LC-2030高效液相色譜儀。色譜條件:色譜柱為IC柱(250×4.6mm,5μm),色譜柱柱溫為25℃;采用紫外檢測器,檢測波長設置為300nm;流動相流速為1.0ml/min;以正己烷-異丙醇-甲醇(330:15:15)為流動相A,以二氯甲烷為流動相B,洗脫條件如下表3所示:表3實施例1的洗脫條件實驗步驟:取替格瑞洛樣品適量,加無水乙醇溶解并稀釋制成每1ml中約含1mg替格瑞洛的溶液,作為供試品溶液;量取供試品溶液適量,用無水乙醇稀釋制成每1ml中約含1μg替格瑞洛的溶液作為對照溶液;另稱取替格瑞洛手性異構體對照品:異構體A+異構體B、異構體C+異構體D、異構體E、異構體F和異構體G各適量,分別加入無水乙醇溶解并稀釋制成一定濃度的貯備液,另稱取替格瑞洛對照品適量,精密加入上述各雜質對照品的貯備液適量,用無水乙醇溶解并稀釋制成每1ml中約含替格瑞洛1mg、各異構體2μg的溶液,作為系統適用性溶液。分別取各溶液10μl并注入液相色譜儀,采用上述色譜檢測條件檢測,記錄色譜圖。圖1示出了實施例1中采用本發明的檢測方法對系統適用性溶液檢測得到的HPLC圖,圖2示出了實施例1中采用本發明的檢測方法對供試品溶液檢測得到的HPLC圖。如圖1和圖2所示,在本發明的色譜條件下替格瑞洛和各手性異構體能有效分離且相鄰手性異構體的最小分離度為1.64,替格瑞洛中無其它手性異構體雜質干擾檢測。對比例1:檢測儀器:島津LC-2030高效液相色譜儀。色譜條件:色譜柱為AD-H柱(250×4.6mm,5μm);以正己烷-無水乙醇(80:20)為流動相;色譜柱柱溫為30℃;采用紫外檢測器;檢測波長設置為300nm;流動相流速為1.0ml/min。實驗步驟:按實施步驟1方法配制系統適用性溶液。取系統適用性溶液10μl,注入液相色譜儀,記錄色譜圖3。由色譜圖3可以看出,在該色譜條件下替格瑞洛和相鄰的手性異構體分離度只有1.15,并且有異構體和主峰位置重疊且平衡時間較長。對比例2:檢測儀器:島津LC-2030高效液相色譜儀。色譜條件:色譜柱為IC柱(250×4.6mm,5μm),色譜柱柱溫為21℃;采用紫外檢測器,檢測波長設置為300nm;流動相流速為1.0ml/min;以正己烷-無水乙醇-異丙醇(85:10:5)為流動相;實驗步驟:按實施步驟1方法配制系統適用性溶液。取系統適用性溶液10μl,注入液相色譜儀,記錄色譜圖4。由色譜圖4可以看出,在該色譜條件下替格瑞洛出峰處有異構體未被分離,相鄰異構體分離度只有1.18。對比例3:檢測儀器:島津LC-2030高效液相色譜儀。色譜條件:色譜柱為IC柱(250×4.6mm,5μm),色譜柱柱溫為30℃;采用紫外檢測器,檢測波長設置為300nm;流動相流速為1.0ml/min;以正己烷-異丙醇-甲醇(330:15:15)為流動相A,以二氯甲烷為流動相B,洗脫條件如下表4所示:表4對比例3的洗脫條件實驗步驟:按實施步驟1方法配制系統適用性溶液。取系統適用性溶液10μl,注入液相色譜儀,記錄色譜圖5,由色譜圖5可以看出,在該色譜條件下替格瑞洛峰型變差、響應變低,并且其與相鄰手性異構體的分離度只有1.157,未達到1.5的分離度。對比例4:檢測儀器:島津LC-2030高效液相色譜儀。色譜條件:色譜柱為IC柱(250×4.6mm,5μm),色譜柱柱溫為30℃;采用紫外檢測器,檢測波長設置為300nm;流動相流速為1.0ml/min;以無水乙醇為流動相A,以二氯甲烷為流動相B,洗脫條件如下表5所示:表5對比例4的洗脫條件時間(min)流動相A(%)流動相B(%)0955608020實驗步驟:按實施步驟1方法配制系統適用性溶液。取系統適用性溶液10μl,注入液相色譜儀,記錄色譜圖6,從色譜圖6可以看出在該色譜條件下,替格瑞洛的保留時間太短且僅為3.818,并且其與手性異構體雜質不能實現有效分離。為了進一步說明本發明的有益效果,本發明提供以下試驗例。試驗例主要用于進行本發明檢測方法的方法學研究,其中,本試驗例中各種試驗均采用如下條件:色譜柱:IC,250×4.6mm,5μm;色譜柱柱溫為30℃;采用紫外檢測器,檢測波長設置為300nm;流動相流速為1.0ml/min;以無水乙醇為流動相A,以二氯甲烷為流動相B,洗脫條件如下表6所示:表6試驗例的洗脫條件時間(min)流動相A(%)流動相B(%)093720861440703045703045.0193760937溶劑:無水乙醇;進樣體積:10μl。專屬性試驗取替羅瑞洛樣品進行酸(0.1mol/L2天)、堿(0.1mol/L2天)、氧化(3%雙氧水2天)、強光(5000LX30天)、紫外光(30天)、高溫(60℃下30天)、高濕(92.5%下30天)等強制降解試驗后用無水乙醇配制成每1ml中約1mg替格瑞洛的供試品溶液。精密量取10μl,注入液相色譜儀,記錄色譜圖。結果見下表7所示:表7試驗例中專屬性試驗結果表7的試驗結果表明,樣品破壞后未產生干擾手性異構體檢測的雜質出現,破壞后產生的異構體能有效地在本發明的檢測條件下檢出。綜上所述,本發明采用高效液相色譜法并利用手性色譜柱對替格瑞洛手性異構體進行分離,式1中替格瑞洛的化合物色譜峰能有效的和其七種手性異構體實現分離,而且采用自身對照法進行雜質的定量測定,操作簡便、檢測成本低且具有良好的靈敏度、線性、專屬性、精密度、準確度、穩定性和耐用性,是控制替格瑞洛異構體的一種行之有效的檢測方法,從而保證了替格瑞洛產品質量和患者用藥安全。本發明并不局限于前述的具體實施方式。本發明擴展到任何在本說明書中披露的新特征或任何新的組合,以及披露的任一新的方法或過程的步驟或任何新的組合。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1