一種長春西汀注射液中維生素C含量的檢測方法與流程
本發明涉及分析化學的
技術領域:
,具體地指一種長春西汀注射液中維生素C含量的檢測方法。
背景技術:
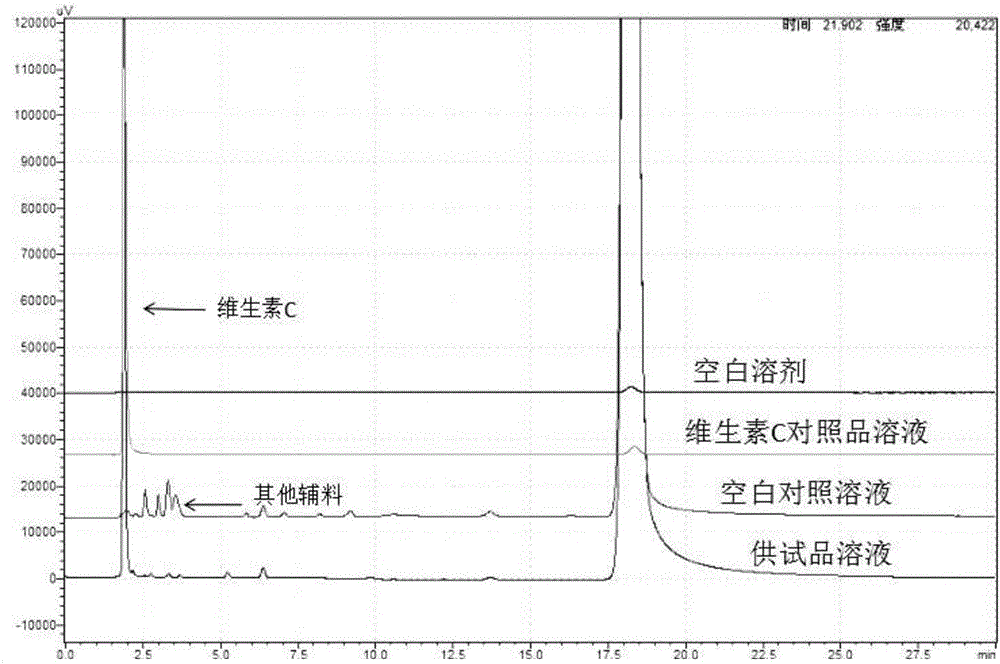
:長春西汀是從夾竹桃科小蔓長春花提取的一種生物堿的衍生物,其有效成分為阿樸長春胺酸乙酯,長春西汀最早由匈牙利的GedeonRichter藥物公司研發并于1978年上市,長春西汀注射液上市以來,因臨床效果顯著以及安全性和耐受性良好,廣泛應用于神經內科、心血管內科、耳科、眼科、神經外科等臨床科室,現已成為世界各國腦血管常規用藥。長春西汀具有多種作用,能改善大腦代謝、血流量以及血液流變學性質。目前上市的長春西汀注射液中輔料一般為維生素C、焦亞硫酸鈉、L-酒石酸、山梨醇、氫氧化鈉和注射用水。維生素C作為長春西汀注射液中重要輔料,起到助溶劑和抗氧劑的作用,它能夠延緩主成分長春西汀的氧化過程,提高產品的穩定性。但維生素C性質不穩定,在高溫條件下易降解,發生氧化反應后容易使溶液顏色變黃,影響產品質量,因此它在長春西汀注射液中的含量變化也應成為該藥品的關注重點。目前,《中華人民共和國藥典》2015版二部收載的維生素C注射液中維生素C含量的檢測方法為滴定法,長春西汀注射液中含有抗氧劑焦亞硫酸鈉,對檢測結果存在干擾。為此,需要研究簡便高效的檢測長春西汀注射液中維生素C含量的檢測方法,以確保藥品的安全性。技術實現要素:本發明的目的就是要提供一種長春西汀注射液中維生素C含量的檢測方法,該方法采用HPLC法檢測長春西汀注射液中維生素C的含量,檢測方法簡便易行,專屬性強、準確度高,為保證藥品的安全、有效、可控提供依據。為實現上述目的,本發明所提供的一種長春西汀注射液中維生素C含量的檢測方法,先通過紫外-可見分光光度計進行光譜掃描確定維生素C標準溶液的最大吸收波長λmax;接著配制一組不同濃度的維生素C溶液,在最大吸收波長λmax下通過高效液相色譜儀測定各維生素C溶液的色譜峰面積,繪制維生素C溶液濃度與其對應色譜峰面積的標準曲線,求得線性回歸方程;同時在最大吸收波長λmax下通過高效液相色譜儀測定供試品溶液的色譜峰面積,最后計算出供試品溶液中維生素C含量。進一步地,所述確定維生素C標準溶液的最大吸收波長λmax的具體步驟如下:取適量維生素C,加入流動相溶解并定量稀釋至濃度為10μg/mL,通過紫外-可見分光光度計在200~400nm范圍內進行光譜掃描,確定最大吸收波長λmax。進一步地,所述繪制維生素C溶液濃度與其對應色譜峰面積A的標準曲線具體步驟如下:配制濃度為2.5μg/ml、12.5μg/ml、25.0μg/ml、37.5μg/ml、50.0μg/ml、125.0μg/ml的維生素C溶液,分別取各溶液10μL進樣,在最大吸收波長λmax下通過高效液相色譜儀測定各維生素C溶液的色譜峰面積,以維生素C的濃度為橫坐標,以色譜峰面積為縱坐標繪制標準曲線,求得到線性回歸方程。進一步地,所述供試品溶液中的維生素C含量測定的具體步驟如下:1)稱取適量待測長春西汀注射液制劑,制成濃度為25μg/mL的供試品溶液;2)精確稱取適量純維生素C,制成濃度為25μg/mL的對照品溶液;3)分別供試品溶液和對照品溶液取15μl注入高效液相色譜儀中,在最大吸收波長λmax下通過測定供試品溶液和對照品溶液的色譜峰面積;4)根據如下公式計算出供試品溶液中維生素C的含量:式中,C為供試品溶液中維生素C的含量,單位為μg/ml;Ai為供試品溶液的峰面積;Ar為對照品溶液的峰面積;Cr為對照品溶液的濃度,單位為μg/ml。進一步地,所述最大吸收波長λmax為265~282nm。最佳地,所述最大吸收波長λmax為267nm。進一步地,所述高效液相色譜儀測定中色譜柱為InertSustainC18柱(4.6mm×250mm,5μm),以十八烷基硅烷鍵合硅膠為填充劑。進一步地,所述高效液相色譜儀測定中柱溫為25~35℃。進一步地,所述高效液相色譜儀測定中流動相為醋酸銨溶液與乙腈按體積比為35:65~45:55的混合液,所述醋酸銨溶液的濃度為0.2mol/L,流速為1ml/min。進一步地,所述高效液相色譜儀測定中定量方法采用外標法。與現有技術相比,本發明具有如下優點:其一,本發明采用HPLC法檢測長春西汀注射液中維生素C的含量,所述流動相與長春西汀注射液有關物質檢測用流動相相同,焦亞硫酸鈉等其他輔料在此檢測條件下無吸收,檢測方法簡便易行,專屬性強、準確度高,為保證藥品的安全、有效、可控提供依據。其二,本發明的測定方法耐用性好,波長、流速等的微小變化對含量測定影響不大,根據長春西汀注射液中各組分在色譜柱固定相和流動相之間分配系數的差別,不同色譜柱均能使維生素C色譜峰與其他色譜峰之間達到較好的分離。其三,在本方法中維生素C經氧化破壞后的降解產物保留時間之前的峰為維生素C降解峰,可見降解產物峰與主峰的分離度良好,不干擾樣品的測定,專屬性好,但維生素C穩定性較差,高溫、堿、氧化等條件都會導致其降解。其四,本發明的方法簡便易行,結果重現性好,靈敏度高,檢測限為0.0301ng;加標回收率穩定,回收率在100.6%~102.92之間,可為維生素C在長春西汀注射液中的含量變化提供關鍵的技術支持。附圖說明圖1為本發明實施例1的紫外-可見光譜掃描示意圖;圖2為本發明實施例2的維生素C的標準曲線;圖3為本發明實施例4中專屬性實驗的測試結果圖。具體實施方式下面結合具體實施例對本發明作進一步的詳細說明。實施例1:本發明確定維生素C標準溶液的最大吸收波長λmax的具體步驟如下:取適量維生素C,加入流動相溶解并定量稀釋至濃度為10μg/mL,通過紫外-可見分光光度計在200~400nm范圍內進行光譜掃描,結果見附圖1所示,維生素C在波長267nm處具有最大吸收波峰。實施例2:本發明繪制維生素C溶液濃度與其對應色譜峰面積A的標準曲線具體步驟如下:精密稱取維生素C配制成濃度為2.5μg/ml、12.5μg/ml、25.0μg/ml、37.5μg/ml、50.0μg/ml、125.0μg/ml的維生素C溶液,分別取各待測試樣溶液10μl進樣,在最大吸收波長λmax下通過高效液相色譜儀測定各維生素C溶液的色譜峰面積,各濃度維生素C溶液濃度(μg/ml)與峰面積見表1。高效液相色譜儀的色譜條件:色譜柱為InertSustainC18柱(4.6mm×250mm,5μm),以十八烷基硅烷鍵合硅膠為填充劑;最大吸收波長λmax為267nm;柱溫為30℃;流動相為醋酸銨溶液與乙腈按體積比為40:60的混合液,所述醋酸銨溶液的濃度為0.2mol/L,流速為1ml/min。表1線性關系考察結果序號123456濃度C(μg/ml)2.512.525.037.550.0125.0峰面積A93172475653994835140348118007054537898以維生素C的濃度為橫坐標,以色譜峰面積為縱坐標繪制標準曲線,如圖2所示,以峰面積A對濃度C作線性回歸,得線性回歸方程:A=36041C+34228,r=0.9997,結果表明在2.5~125.0μg/ml范圍內,維生素C對照品的濃度與峰面積A線性關系良好。實施例3:本發明測定長春西汀注射液中維生素C含量的具體步驟:1)稱取適量樣品1~3待測長春西汀注射液制劑,制成濃度為25μg/mL的供試品溶液;2)精確稱取適量純維生素C,制成濃度為25μg/mL的對照品溶液;3)分別供試品溶液和對照品溶液取15μl注入高效液相色譜儀中,在最大吸收波長λmax下通過測定供試品溶液和對照品溶液的色譜峰面積;4)根據如下公式計算出供試品溶液中維生素C的含量:式中,C為供試品溶液中維生素C的含量,單位為μg/ml;Ai為供試品溶液的峰面積;Ar為對照品溶液的峰面積;Cr為對照品溶液的濃度,單位為μg/ml。高效液相色譜儀的色譜條件:色譜柱為InertSustainC18柱(4.6mm×250mm,5μm),以十八烷基硅烷鍵合硅膠為填充劑;最大吸收波長λmax為267nm;柱溫為30℃;流動相為醋酸銨溶液與乙腈按體積比為40:60的混合液,所述醋酸銨溶液的濃度為0.2mol/L,流速為1ml/min。樣品1-3檢測所用的長春西汀注射液成分為長春西汀、維生素C、焦亞硫酸鈉、山梨醇、L-酒石酸、氫氧化鈉和注射用水,其中維生素C理論值含量為0.25mg/ml,每個實施例中維生素C含量檢測進行2次平行實驗,結果如表2所示。表2維生素C含量檢測結果樣品1-3中檢測結果與理論值0.25mg/ml接近,偏低的原因是由于在生產和儲存過程中會消耗部分維生素C。實施例4:本發明測定方法的專屬性實驗:在實施例3中的色譜條件下,精密量取上述對照品溶液、待測試樣溶液、空白溶劑、無維生素C含長春西汀和其他輔料的空白對照溶液各10μl,分別注入液相色譜儀,記錄色譜圖,對色譜圖中的主要成分峰進行歸屬并考察選擇性,結果如圖3所示。對照品溶液色譜圖中,維生素C保留時間為1.889分鐘;待測試樣溶液色譜圖中,維生素C峰與其他色譜峰分離度良好;空白對照溶液在維生素C峰相應保留時間處無干擾吸收峰,對含量測定無干擾。實施例5本發明測定方法的精密度試驗:1、儀器精密度試驗:對照品溶液(濃度為25.0μg/ml),按上述色譜條件重復進樣6次,測定峰面積,結果見表3。表3儀器精密度試驗結果*rsd(%)=標準偏差sd/平均值×100%結果表明,其峰面積測量值的RSD為1.79%,說明儀器精密度良好。2、重復性試驗:取本品10支,混合均勻后按“2.3”項下方法平行制備6份待測試樣溶液,測定峰面積,按外標法計算本品中維生素C的濃度,結果見表4。表4重復性試驗結果(μg/ml)序號123456平均值RSD濃度150.4151.9146.0150.1150.4151.1150.01.37%結果表明,本品中維生素C的濃度為150.0μg/ml,測量結果的RSD小于2.0%,說明該方法重復性良好。3、中間精密度試驗:取本品10支,混合均勻后由不同的操作人員按“2.3”項下方法平行制備6份待測試樣溶液,測定峰面積,按外標法計算本品中維生素C的濃度,結果見表5。表5中間精密度試驗結果(μg/ml)結果表明,不同操作人員測定本品中維生素C濃度為178.8μg/ml,測量結果的RSD小于2.0%,說明該方法中間精密度良好。實施例6本發明測定方法的準確度試驗:取本品10支,混合均勻后量取0.5ml于10ml量瓶中,按“實施例3”項下待測試樣溶液80%~120%取樣量的范圍取樣,分別加入對照品溶液6ml、5ml和4ml,用流動相稀釋至刻度,搖勻,即平行制備高濃度、中濃度、低濃度待測試樣溶液各三份。按上述色譜條件,分別取待測試樣溶液和對照品溶液各10μl,進樣,記錄色譜圖,測定峰面積。按外標法計算維生素C的含量,結果見表6。表6維生素C回收率測定結果可見在80%~120%測試濃度的范圍內,測定結果的準確度良好。實施例7本發明測定方法的范圍:配制三種規格待測試樣中維生素濃度分別25μg/ml、41.7μg/ml和50μg/ml,選擇最低濃度的80%(20μg/ml)作為低濃度溶液,最高濃度的120%(60μg/ml)作為高濃度溶液,取維生素C對照品適量,平行制備各濃度待測試樣溶液三份,精密量取10μl進樣,測定峰面積,按外標法計算各待測試樣溶液中維生素C的濃度,結果見表7。表7維生素C范圍試驗結果結果表明,維生素C可以在20μg/ml~60μg/ml范圍內取樣定量。實施例8本發明測定方法的耐用性試驗1、不同檢測波長的比較考察檢測波長的改變對含量測定結果的影響。使用不同的檢測波長(±2nm),其他色譜條件不變,分別精密量取待測試樣溶液及對照品溶液各10μl,注入液相色譜儀,測定待測試樣溶液中維生素C的峰面積,按外標法計算本品中維生素C的濃度,結果見表8。表8耐用性試驗-不同檢測波長的比較檢測波長267nm278nm280nm282nmRSD濃度(μg/ml)179.7174.0174.6171.51.97%結果顯示:本品中維生素C濃度的RSD小于2.0%,表明在一定范圍內,不同的檢測波長對含量測定結果無影響。但因維生素C在波長267nm處具有最大吸收,故最終確定選擇267nm作為本方法的檢測波長。2、不同流速的比較考察流速的改變對含量測定結果的影響。使用不同的流速(±0.1ml/min),其他色譜條件不變,分別精密量取待測試樣溶液及對照品溶液各10μl,注入液相色譜儀,測定待測試樣溶液中維生素C的峰面積,按外標法計算本品中維生素C的濃度,結果見表9。表9耐用性試驗-不同流速的比較流速0.9ml/min1.0ml/min1.1ml/minRSD濃度(μg/ml)169.9174.6173.51.40%結果顯示:本品中維生素C濃度的RSD小于2.0%,表明在一定范圍內,不同的流速對含量測定結果無影響。3、不同流動相比例考察不同流動相比例對含量測定結果的影響。使用不同流動相比例,其他色譜條件不變,分別精密量取待測試樣溶液及對照品溶液各10μl,注入液相色譜儀,測定待測試樣溶液中維生素C的峰面積,按外標法計算本品中維生素C的濃度,結果見表10。表10耐用性試驗-不同流動相比例的比較乙腈:0.2mol/l醋酸銨溶液65:3560:4055:45RSD濃度(μg/ml)171.2173.0173.20.64%結果顯示:本品中維生素C濃度的RSD小于2.0%,表明在一定范圍內,不同的流動相比例均可達到較好的分離,對其含量測定無影響。結論:該檢測方法耐用性良好。以上所述,僅為本發明的具體實施方式,應當指出,任何熟悉本領域的技術人員在本發明所揭露的技術范圍內,可輕易想到的變化或替換,都應涵蓋在本發明的保護范圍之內。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1