西紅花色香味多組分定量結合指紋圖譜質量評價方法與流程
本發明涉及中藥活性成分分析技術領域,具體地說,是涉及一種西紅花色香味多組分定量結合指紋圖譜質量評價方法。
背景技術:
西紅花(Crocus sativus L.)又稱藏紅花,以雌蕊上部三根紅色柱頭入藥,具有獨特的顏色、香氣和苦味,價格昂貴,為名貴中藥,也是世界上最名貴香料,目前從西紅花中分離得到的化合物已達到50余種,主要可分為二萜類(以西紅花苷類化合物為代表,為西紅花紅色的主要成分)、單萜類(以苦藏花素和藏花醛為代表,分別是西紅花的苦味和香氣成分)、黃酮類(以山奈酚為苷元),其中西紅花苷中的西紅花苷-Ⅰ、西紅花苷-Ⅱ和苦藏花素含量較高,三者之和可占干重的40%以上,其它化合物的質量比例都較小。作為食品添加劑的國際標準ISO 3632-2(2010版),用分光光度法分別于λmax440nm、λmax 330nm和λmax 257nm測定西紅花水提取液中色、香、味的代表成分西紅花苷、藏花醛和苦藏花素的色價來進行質量控制與分級。規定西紅花苷色價大于190,苦藏花素色價大于70,藏花醛色價介于20-50之間為一級品。但該方法專屬性不強,不能精確表示色、香、味三類組分的含量,如西紅花苷類,在257nm和330nm處也有吸收,存在干擾。
2015年版《中國藥典》用HPLC法測定西紅花苷-Ⅰ和西紅花苷-Ⅱ的含量,規定兩者總含量大于10%為合格品,該標準只對西紅花苷中西紅花苷-Ⅰ和西紅花苷-Ⅱ進行含量控制,對其他西紅花苷、苦藏花素、藏花醛等則沒有規定。苦藏花素和藏花醛均具有較強的藥理活性,為西紅花的藥效成分,并且苦藏花素在西紅花中含量均較高,苦藏花素納入西紅花質量控制指標能更有效地控制和評價西紅花藥材的質量。
定量指紋圖譜技術是以指紋圖譜與多成分定量相結合的綜合評價方法,能較為全面地反映中藥及其制劑中所含化學成分的種類與數量,且能夠同時進行多種藥效成分的定量分析,進而可以對藥品質量進行整體描述和評價,體現出中藥的整體觀。文獻報道,不同產地,不同質量等級的西紅花所含化學成分的種類與數量有較大的差異,使用定量指紋圖譜技術更能全面反映西紅花藥材質量,為臨床安全、合理用藥提供依據。
技術實現要素:
本發明所要解決的技術問題是提供一種測定西紅花提取液中色、香、味的代表成分西紅花苷、藏花醛和苦藏花素等多組份含量;對其他復雜成分利用指紋圖譜進行質量控制的方法。
為解決上述技術問題,本發明提供了一種西紅花色香味多組分定量結合指紋圖譜質量評價方法,包括步驟:
a.確定高效液相色譜條件;
b.確定質譜條件;
c.制備供試品溶液;
d.制備色味混合對照品和藏花醛對照品溶液;
e.取對照品和西紅花樣品溶液進樣分析,得對照品和西紅花樣品高效液相色譜圖和質譜總離子流色譜圖,首先基于液相色譜的在線紫外圖譜判斷化合物的結構類型,再利用波峰在正負離子模式下離子裂解規律推斷化合物的結構;
f.光譜和質譜分析西紅花中的色譜峰特征;
g.建立西紅花定量指紋圖譜方法:
精密度試驗:取同一供試品溶液按指紋圖譜的色譜條件,連續進樣6次,采用國家藥典委員會中藥色譜指紋圖譜相似度評價系統2004A版計算其相似度;
穩定性試驗:取同一供試品溶液,在步驟b中的色譜條件下分別在0、2、4、8、12、20、24h測定,記錄色譜圖,測定樣品穩定性;
重復性試驗:取西紅花樣品6份,精密稱定,按照步驟c中的方法制備供試品溶液,按照步驟a中的液相色譜條件下進行測定,記錄色譜圖,考察實驗方法的重復性;
h.西紅花樣品波長切換HPLC指紋圖譜的建立和定量:取干品西紅花,按步驟供試品溶液制備方法和檢測方法進行檢測,得到樣品的高效液相色譜圖,將西紅花樣品的高效液相色譜圖譜信號數據導入國家藥典委員會中藥指紋圖譜相似度評價系統(2004A版)軟件進行處理,設定第一個樣品為參照圖譜,選取時間窗寬度為0.1min,對照圖譜的生成方法為中位數,選擇穩定性和重復性在0.999以上、吸收較強、特征明顯的色譜峰為共有特征峰,進行多點校正,自動匹配,生成對照圖譜,11個共有色譜峰為:苦藏花素、槐糖苷、trans-5-tG、西紅花苷-Ⅰ、西紅花苷-Ⅱ、trans-2-G、藏花醛、西紅花苷-Ⅰ的順式異構體、西紅花苷-Ⅱ的順式異構體以及trans-1-g;然后進行各樣品的相似度評價,以供試品溶液指紋圖譜中西紅花苷-Ⅰ的保留時間及峰面積來計算其他共有峰相對保留時間和相對峰面積,并統計結果。
優選地:所述步驟a中,高效液相色譜條件:Ultimate XB C18色譜柱;柱溫:30℃;流動相:0.4%甲酸-甲醇,梯度洗脫,0-60min,甲醇濃度:20%-100%;DAD波長范圍200~600nm,檢測時間在25min內測定波長為260nm,25-29min之間測定波長為440nm,29-36min之間測定波長為260nm,36-40.5min之間測定波長為440nm,40.5-42.5min之間測定波長為310nm,42.5-60min之間測定波長為440nm;流速:0.8ml·min-1;進樣量:10μl。
優選地:所述步驟b中,所述質譜條件進一步為,離子源:大氣壓電噴霧離子源ESI;檢測模式:正負離子;全掃描質核比:50-1200m/z;干燥氣溫度:350℃,干燥氣流量:12.0L·min-1,霧化氣壓力:35.0psi;毛細管電壓:4.5kV;碰撞電壓1.0V;進入質譜的流動相流速分流至0.4ml·min-1。
優選地,所述步驟c,進一步為,稱取西紅花樣品粉末10mg,精密稱定,置25ml棕色量瓶中,加甲醇20ml,冰浴超聲處理30min,放置室溫,加甲醇定容至刻度,搖勻,0.22μm微孔濾膜濾過,取續濾液,得到供試品溶液。
優選地,其特征在于,所述步驟d,進一步為,
1)制備色味混合對照品溶液:精密稱取西紅花苷-Ⅰ對照品、西紅花苷-Ⅱ對照品、以及苦藏花素置于棕色量瓶中,用甲醇溶解并定容至刻度。得濃度為59.6μg·mL-1、29.6μg·mL-1和40.8μg·mL-1的西紅花苷-Ⅰ、西紅花苷-Ⅱ和苦藏花素混合對照品溶液,搖勻,備用;
2)制備藏花醛對照品溶液:精密吸取藏花醛對照品51.5mg,置25mL 棕色量瓶中,用甲醇溶解并定容至刻度,得濃度為2.06mg·mL-1對照品溶液,作為儲備液。從中精密吸取0.5mL,置25mL棕色量瓶中,加甲醇稀釋至刻度,得濃度為41.2μg·mL-1對照品溶液。
優選地,其特征在于,所述步驟e中,進一步為,采用波長切換方法在440nm、250nm和330nm下測定西紅花中的西紅花苷、苦藏花素和藏花醛化合物,得對照品和西紅花樣品高效液相色譜圖和質譜總離子流色譜圖。
西紅花的國際標準ISO 3632-2,用分光光度法測定西紅花的色價來進行質量控制與分級,該方法專屬性不強,不能精確表示色、香、味三類組分的含量。《中國藥典》用HPLC法測定,但該標準只對西紅花苷中西紅花苷-Ⅰ和西紅花苷-Ⅱ進行含量控制,對其他藥效成分如苦藏花素、藏花醛等則沒有規定。本發明首先采用HPLC-DAD-ESI-MSn技術對西紅花中主要化學成分進行分析和鑒別,在基于西紅花中“色香味”多藥效組分和質譜鑒定的基礎上,利用波長切換HPLC-DAD技術通過色-香-味多組分定量結合指紋圖譜,構建了西紅花的多組分質量評價體系,可對西紅花進行較全面的質量分析,可提高西紅花臨床應用的安全性和有效性。
與現有技術相比,本發明所述的西紅花色香味多組分定量結合指紋圖譜質量評價方法,達到了如下效果:
國際標準ISO 3632-2,用分光光度法測定西紅花水提取液中色、香、味的代表成分西紅花苷、藏花醛和苦藏花素的色價來進行質量控制與分級。但該方法專屬性不強,不能精確表示色、香、味三類組分的含量。《中國藥典》用HPLC法測定,但該標準只對西紅花苷中西紅花苷-Ⅰ和西紅花苷-Ⅱ進行含量控制,對其他藥效成分如苦藏花素、藏花醛等則沒有規定,因此現行國際標準與中國藥典對西紅花質量評價尚存在較大的缺陷。
本發明首先采用HPLC-DAD-ESI-MSn技術對西紅花中主要化學成分進行分析和鑒別,在基于西紅花中“色香味”多藥效組分和質譜鑒定的基礎上,利用波長切換HPLC-DAD技術通過色-香-味多組分定量結合指紋圖譜,構建了西紅花的多組分質量評價體系,可對西紅花的14種成分進行定性與定量分析,測定結果穩定可靠,比現有方法更全面評價西紅花質量,可提高西紅花臨床應用的安全性和有效性。
本發明采用HPLC-DAD-ESI-MSn技術對西紅花中主要化學成分進行分析和鑒別,在基于西紅花中“色香味”多藥效組分和質譜鑒定的基礎上,利用波長切換HPLC-DAD技術通過色-香-味多組分定量結合指紋圖譜,構建了西紅花的多組分質量評價體系,可對西紅花進行較全面的質量分析,可提高西紅花臨床應用的安全性和有效性。
附圖說明
此處所說明的附圖用來提供對本發明的進一步理解,構成本發明的一部分,本發明的示意性實施例及其說明用于解釋本發明,并不構成對本發明的不當限定。在附圖中:
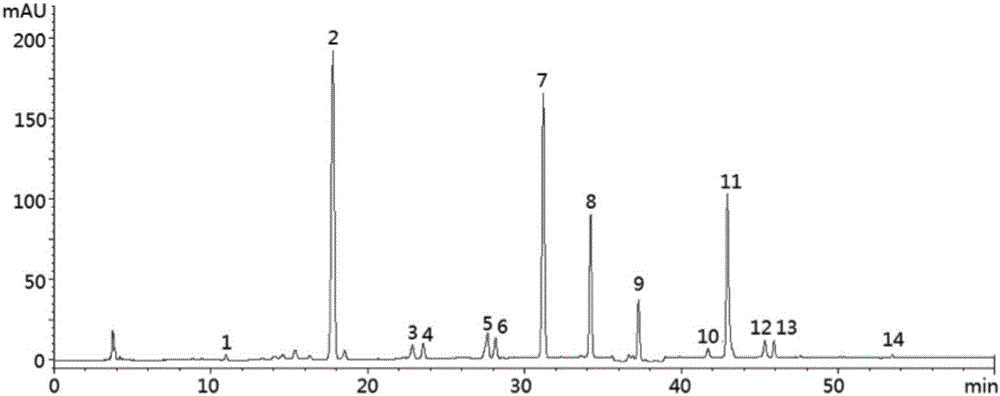
圖1為實施例1中對照品和樣品波長切換液相色譜圖;
圖2a-圖2b為西紅花總離子流圖,圖2a為正離子模式,圖2b為負離子模式;
圖3為實施例1中的精密度試驗色譜圖;
圖4為實施例1中的穩定性試驗色譜圖;
圖5為不同提取溶劑液相色譜HPLC圖;
圖6為超聲提取不同時間HPLC圖;
圖7為對照圖譜;
圖8a-圖8c為28批西紅花樣品的共有模式圖,其中s1-s28分別代表樣品1-樣品28;
圖9a-圖9c為西紅花對照品和樣品波長切換HPLC圖。
具體實施方式
如在說明書及權利要求當中使用了某些詞匯來指稱特定組件。本領域技術人員應可理解,硬件制造商可能會用不同名詞來稱呼同一個組件。本說明書及權利要求并不以名稱的差異來作為區分組件的方式,而是以組件在功能上的差異來作為區分的準則。如在通篇說明書及權利要求當中所提及的“包含”為一開放式用語,故應解釋成“包含但不限定于”。“大致”是指在可接收的誤差范圍內,本領域技術人員能夠在一定誤差范圍內解決所述技術問題,基本達到所述技術效果。說明書后續描述為實施本發明的較佳實施方式,然所述描述乃以說明本發明的一般原則為目的,并非用以限定本發明的范圍。本發明的保護范圍當視所附權利要求所界定者為準。
實施例1:
本實施例提供了一種西紅花色香味多組分定量結合指紋圖譜質量評價方法,具體步驟如下:
A.高效液相色譜條件:Ultimate XB C18色譜柱(250mm×4.6mm,5μm);柱溫:30℃;流動相:0.4%甲酸(A)-甲醇(B),梯度洗脫,0-60min(B:20%-100%);DAD波長范圍200~600nm,0-25min(260nm),25-29min(440nm),29-36min(260nm),36-40.5min(440nm),40.5-42.5min(310nm),42.5-60min(440nm);流速:0.8ml·min-1;進樣量:10μL。
B.質譜條件:離子源:大氣壓電噴霧離子源(ESI);檢測模式:正負離子;全掃描質核比(m/z):50-1200;干燥氣溫度:350℃,干燥氣流量:12.0L·min-1,霧化氣壓力:35.0psi;毛細管電壓:4.5kV;碰撞電壓1.0V;進入質譜的流動相流速分流至0.4ml·min-1。
C.供試品溶液制備
稱取西紅花樣品粉末約10mg,精密稱定,置25ml棕色量瓶中,加甲醇20mL,冰浴超聲處理30min,放置室溫,加甲醇定容至刻度,搖勻,0.22μm微孔濾膜濾過,取續濾液,即得。
D.對照品溶液制備
1)色-味混合對照品溶液制備:精密稱取西紅花苷-Ⅰ對照品2.98mg,西紅花苷-Ⅱ對照品1.48mg,苦藏花素2.02mg置50ml棕色量瓶中,用甲醇溶解并定容至刻度。得濃度為59.6μg·ml-1、29.6μg·ml-1和40.8μg·ml-1的西紅花苷-Ⅰ、西紅花苷-Ⅱ和苦藏花素混合對照品溶液,搖勻,備用。
2)藏花醛對照品溶液制備:精密吸取藏花醛對照品51.5mg,置25ml棕色量瓶中,用甲醇溶解并定容至刻度,得濃度為2.06mg·ml-1對照品溶液,作為儲備液。從中精密吸取0.5ml,置25ml棕色量瓶中,加甲醇稀釋至刻度,得濃度為41.2μg·ml-1對照品溶液。
E.液相色譜圖和總離子流圖的建立
通過對流動相和檢測波長考察,建立了較佳的液相色譜分析條件,基本實現了西紅花中主要化合物的基線分離。由于西紅花中的西紅花苷、苦藏花素和藏花醛三類主要化合物在440nm、250nm和330nm下有特征性吸收,但吸收強度相差很大,所以本發明采用了波長切換技術。按上述實驗條件,取對照品和西紅花樣品溶液進樣分析,得對照品和西紅花樣品高效液相色譜圖(圖1)和質譜總離子流色譜圖(圖2a和圖2b)。
從HPLC圖和總離子流圖可看出,液相上的色譜峰基本均有質譜響應,正離子模式比負離子模式色譜峰多,并且響應峰的強度大,但有的色譜峰在負離子下有響應的,正離子無響應。所以本發明首先基于HPLC的在線紫外圖譜,判斷化合物的可能結構類型,再進一步利用各峰在正負離子模式下離子裂解規律推斷化合物的結構。
F.西紅花中14個色譜峰光譜和質譜特征分析
由于流動相中存在鈉離子和甲酸根離子,通過正負離子模式下的總離子流圖可發現,西紅花中多種西紅花苷類成分、苦藏花素和黃酮類成分在正離子模式下主要產生[M+Na]+峰,在負離子模式下主要產生[M-H]-和[M+HCOO]-峰。為了更好地歸納分析西紅花化學成分的質譜裂解規律,通過與對照品保留時間、在線紫外圖譜和化合物的一級、二級、三級質譜裂解特征,并通過對比同類化合物的質譜裂解規律和質譜碎片特征,參考現有技術相關文獻進行推測。結果見表1。
表1 14個峰UV和質譜裂解規律
G.西紅花定量指紋圖譜方法建立
精密度試驗:取同一供試品溶液按指紋圖譜的色譜條件,連續進樣6次,采用國家藥典委員會“中藥色譜指紋圖譜相似度評價系統2004A版”計算其相似度在0.999以上,表明儀器的精密度良好。見圖3。
穩定性試驗:取同一供試品溶液,在前述色譜條件下分別在0、2、4、8、12、20、24h測定,記錄色譜圖,考察樣品穩定性。經相似度評價,均在0.999以上,表明供試品溶液24h內穩定性良好。見圖4。
重復性試驗:取S15樣品6份,精密稱定,按照“供試品溶液制備”項下的方法制備供試品溶液,在前述色譜條件下分別進行測定,記錄色譜圖,考察實驗方法的重復性。經相似度評價,均在0.999以上,表明方法重復性良好。
H.西紅花樣品波長切換HPLC指紋圖譜的建立和定量
取干品西紅花,按以上供試品溶液制備方法和檢測方法進行檢測,得到樣品的高效液相色譜圖。將樣品HPLC圖譜信號數據導入國家藥典委員會《中藥指紋圖譜相似度評價系統(2004A版)》軟件進行處理,設定第一個樣品為參照圖譜,選取“時間窗寬度”為0.1min,對照圖譜的生成方法為“中位數”,選擇穩定性和重復性好,吸收較強,特征明顯的11個色譜峰為共有特征峰,進行多點校正,自動匹配,生成“對照圖譜”,11個共有色譜峰為:
1:苦藏花素,2:槐糖苷,3:trans-5-tG,4:未鑒定出,5:西紅花苷-Ⅰ,6:西紅花苷-Ⅱ,7:trans-2-G,8:藏花醛,9:西紅花苷-Ⅰ的順式異構體(cis-4-GG),10:西紅花苷-Ⅱ的順式異構體(cis-4-Gg),11:trans-1-g;
然后進行各樣品的相似度評價,以供試品溶液指紋圖譜中西紅花苷-Ⅰ的保留時間及峰面積來計算其他共有峰相對保留時間和相對峰面積,并統計結果。
實例2:西紅花供試液制備方法的優化
1.儀器、試劑和材料
Agilent 1200高效液相色譜儀(美國Agilent公司);Agilent 6330離子阱質譜儀,配有電噴霧離子源(ESI)(美國Agilent公司);AG135電子天平(瑞士Mettle Toledo公司);Milli-Q超純水儀(美國Millipore公司);KQ-500DB型數控超聲波清洗器(昆山市超聲儀器有限公司)。
西紅花苷-Ⅰ(批號:111588-201202),西紅花苷-Ⅱ(批號:111589-201304),供HPLC含量測定用,均購于中國食品藥品檢定研究院;藏花醛(批號:101198434,Sigma公司,含量>88%);苦藏花素(自制,含量>90%);甲醇和甲酸均為色譜純(美國Tedia公司),其余化學試劑均為分析純。
西紅花藥材由湖州西紅花種植基地提供,經浙江中醫藥大學陳錫林教授鑒定為Crocus sativus L.的干燥花柱。研磨后,過60目篩,避光干燥保存。
2.方法
2.1供試品溶液制備
2.1.1提取溶劑選擇
鮮品西紅花柱頭主要含有多種西紅花苷類成分(以西紅花苷-Ⅰ和西紅花苷-Ⅱ為主,占總西紅花苷90%以上)和苦藏花素,兩類成分可占干重的40%以上,極性較大,溶于水、甲醇和乙醇。加熱干燥過程中苦藏花素會部分水解,生成苷元,進一步脫水生成藏花醛,含量較低。藏花醛為西紅花的香氣成分,不溶于水,可溶于甲醇、乙醇、乙醚等。本研究以西紅花中的色-香-味三類成分為指標,對提取溶劑進行考察。見圖5。
結果表明,水對西紅花苷-Ⅰ的提取率最低,并且無藏花醛色譜峰。50%乙醇、70%甲醇和甲醇對苦藏花素和西紅花苷-Ⅰ、Ⅱ提取率基本相似,但甲醇對藏花醛的提取率較高,并且樣品在甲醇中的穩定性最好,故本研究以甲醇為提取溶劑。
2.1.2提取時間
現有技術中的文獻報道,西紅花中的西紅花苷類成分穩定性較差,2015年版《中國藥典》西紅花項下,西紅花苷-Ⅰ和西紅花苷-Ⅱ含量測定,用超聲法提取,并要求在冰浴上操作,本研究為進一步考察超聲法對西紅花苷類成分,苦藏花素和藏花醛的提取率的影響,對超聲時間進行了優化。見圖6。
結果表明,不同超聲提取時間,成分種類基本一致,但提取率有差異。隨著提取時間延長西紅花苷-Ⅰ、西紅花苷-Ⅱ和苦藏花素提取率增加,達到30min時提取率最高,之后變化不大。故超聲提取30min。
2.1.3供試品溶液制備
在優化了提取溶劑和超聲時間的基礎上,最終確定了供試品溶液的制備方法,具體如下:
稱取西紅花樣品粉末約10mg,精密稱定,置25mL棕色量瓶中,加甲醇20mL,冰浴超聲處理30min,放置室溫,加甲醇定容至刻度,搖勻,0.22μm微孔濾膜濾過,取續濾液,即得。
實例3:28批國產西紅花波長切換HPLC指紋圖譜的建立和分析
取不同來源的28批干品西紅花,按以上供試品溶液制備方法和檢測方法進行檢測,得到28批樣品的高效液相色譜圖。將28批樣品HPLC圖譜信號數據導入國家藥典委員會《中藥指紋圖譜相似度評價系統(2004A版)》軟件進行處理,設定S1為參照圖譜,選取“時間窗寬度”為0.1min,對照圖譜的生成方法為“中位數”,選擇穩定性和重復性好,吸收較強,特征明顯的11個色譜峰為共有特征峰,進行多點校正,自動匹配,生成“對照圖譜”,見圖7。11個共有色譜峰為:
1:苦藏花素,2:槐糖苷,3:trans-5-tG,4:未鑒定出,5:西紅花苷-Ⅰ,6:西紅花苷-Ⅱ,7:trans-2-G,8:藏花醛,9:西紅花苷-Ⅰ的順式異構體(cis-4-GG),10:西紅花苷-Ⅱ的順式異構體(cis-4-Gg),11:trans-1-g
匹配后的28批樣品譜圖見圖8a-8c。然后進行各樣品的相似度評價,見表3。除樣品S5、S7和S20三個樣品外,其余25批樣品相似度均在0.98以上,尤其S7相似度最低。從圖8a-8c和表3相似度可知,S5、S7和S20的9號、10號和11號色譜峰響應值較高,從表5相對峰面積可看出,這三批樣品的9號色譜峰相對峰面積在1以上,而其他樣品的該色譜峰的相對峰面積在1以下,可能是引起3批樣品相似度低的主要原因。根據第一章質譜鑒定結果,表明該成分為西紅花苷-Ⅰ的順式異構體(cis-4-GG),后期研究表明,該成分在鮮品中含量較低,其干品中的含量與干燥溫度等有關。并且S7的相似度最低(0.934),從表5可看出,該樣品1號色譜峰(苦藏花素)相對峰面積為1,其他樣品該色譜峰的相對峰面積均大于1,8號色譜峰(藏花醛)和9號色譜峰相對峰面積均較其他樣品高,可能是該樣品相似度低的主要原因。
通過與對照品對照和結合第一章質譜鑒定結果,確定5號峰為西紅花苷-Ⅰ。以5號峰作為參照峰S,以供試品溶液指紋圖譜中西紅花苷-Ⅰ的保留時間及峰面積來計算其他共有峰相對保留時間和相對峰面積,統計結果分別見表4和表5。
表3 28批國產西紅花指紋圖譜相似度
表4 28批西紅花共有色譜峰相對保留時間
表5 28批西紅花共有色譜峰相對峰面積
從相對保留時間和相對峰面積數據可看出,不同來源國產西紅花藥材指紋圖譜中共有峰相對保留時間基本一致(RSD<0.5%);但由于來源不同,可能采后加工方法等不同,各批藥材共有峰的相對峰面積有較大差異(即各峰代表成分含量的相對比值則有較大差別),差異最大的是峰8(藏花醛) 和峰10(cis-4-Gg);其次是峰9(cis-4-GG)和峰11(trans-1-g)。由此可知,建立的色譜方法可以使不同來源西紅花藥材具有基本一致的色譜行為(相對保留時間),但藥材之間西紅花苷類和苦藏花素等成分的含量受多種因素的影響,具有較大差異(相對峰面積)。提示不同產地西紅花存在差異,必須嚴格控制西紅花質量,確保其臨床療效。
實例4:色香味HPLC法同時定量分析國內外西紅花樣品
1.1儀器、試劑和材料
儀器、試劑、材料同實例2
1.2方法
1.2.1供試品溶液制備
同實例2
1.2.2對照品溶液制備
同實例2
1.2.2.1色-味混合對照品溶液制備
同實例2
1.2.2.2藏花醛對照品溶液制備
同實例2
1.3色譜條件
Ultimate XB C18色譜柱(250mm×4.6mm,5μm);柱溫:30℃;流動相:0.4%甲酸(A)-甲醇(B),梯度洗脫,0-60min(B:20%-100%);DAD波長范圍200~600nm,0-25min(260nm),25-29min(440nm),29-36min(260nm),36-40.5min(440nm),40.5-42.5min(310nm),42.5-51min(440nm);流速:0.8mL·min-1;進樣量:10μL。波長切換代表色譜圖見圖9a-9c。
1.4方法:方法于現有技術中的定量指紋圖譜的方法相同
1.4.1線性關系考察
1.4.1.1色味線性關系考察
分別精密吸取西紅花苷-Ⅰ、西紅花苷-Ⅱ和苦藏花素混合對照品溶液1、2、4、6、8、10μL,按上述色譜條件進樣分析,測定峰面積。以峰面積Y為縱坐標,對照品質量為橫坐標(X,μg),繪制標準曲線。得回歸方程和線性范圍,結果見表8,三個成分在各自濃度范圍內線性關系良好。
1.4.1.2香(藏花醛)線性關系考察
分別精密吸取藏花醛對照品溶液1、2、4、6、8、10μL,按上述色譜條件進樣分析,測定峰面積。以峰面積Y為縱坐標,對照品質量為橫坐標(X,μg),繪制標準曲線。得回歸方程和線性范圍,結果見表8,藏花醛在一定濃度范圍內線性關系良好。
表8西紅花中4個成分的線性關系
1.5樣品含量測定
按前述供試品溶液制備方法和色譜條件測定28批國產西紅花中西紅花苷-Ⅰ、西紅花苷-Ⅱ、苦藏花素和藏花醛的含量,結果見表10。
2.UV法測定總西紅花苷含量方法的建立
西紅花中的西紅花苷類成分有其特殊性,苷元均為西紅花酸,紫外吸收光譜特征基本相同,最大吸收波長均為440nm。《中國藥典》方法測定的28批西紅花樣品的結果表明,西紅花苷-Ⅰ含量均比西紅花苷-Ⅱ含量高,基本是西紅花苷-Ⅱ的2倍以上;并且西紅花苷-Ⅱ對照品的價格是西紅花苷-Ⅰ的2倍以上,故本文以西紅花苷-Ⅰ為對照,有其合理性。
2.1對照品溶液的制備
精密稱取西紅花苷-Ⅰ對照品6.20mg,置25mL棕色量瓶中,用甲醇溶解并定容至刻度。從中精密移取2.0mL置10mL量瓶中,用甲醇稀釋至刻度,搖勻,得濃度為49.6μg·mL-1的對照品溶液。
2.2供試品溶液制備
精密吸取“色-香-味HPLC法同時定量分析方法”供試品溶液5mL,置50mL棕色量瓶中,用甲醇稀釋至刻度。
2.3測定波長的選擇
對照品溶液及供試品溶液分別于200-700nm進行波長掃描,發現兩者均在440nm處有最大吸收,故確定檢測波長為440nm。與西紅花苷類成分的最大吸收波長相一致。
2.4線性關系考察
分別精密吸取對照品溶液0.1,0.2,0.4,0.6,0.8,1.0mL置于10mL量瓶中,用甲醇稀釋至刻度,以甲醇作空白對照,照紫外-可見分光光度法,于440nm波長處測定吸光度,得回歸方程為Y=0.1130X+0.0012(r=0.9999)。結果表明,西紅花苷-Ⅰ在0.496-4.96μg·mL-1濃度范圍內與吸光度呈良好線性關系。
2.5精密度和穩定性考察
精密吸取0.6mL對照品溶液,按2.4項下方法操作,重復測定6次,吸光度的RSD為0.032%,說明儀器精密度良好。
取S1樣品液,按2.4項下方法操作,分別在0min、10min、20min、30min、40min、50min、60min測定吸光度,RSD為0.38%,說明樣品在1h內穩定性良好。
2.6重復性考察
取S1樣品,平行6份,精密稱定,分別按2.2和2.4項下方法操作,測定吸光度,按標準曲線分別計算總西紅花苷含量,計算平均含量為27.60%,RSD為1.98%。說明該方法重復性良好。
2.7加樣回收率考察
取已知總西紅花苷含量(27.60%,重復性試驗結果)的S1樣品,平行6份,每份5.0mg,精密稱定,各加入西紅花苷-Ⅰ對照品溶液5mL,分別按2.2和2.4項下方法操作,測定吸光度,按標準曲線分別計算總西紅花苷含量,計算平均回收率,結果見表9。表明該方法準確可靠。
表9西紅花中總西紅花苷加樣回收率試驗結果
2.8UV法樣品中總西紅花苷含量測定
取28批西紅花藥材,分別按“2.2”中的方法制備供試品溶液,按“2.4”中的方法操作,測定吸光度,按標準曲線分別計算總西紅花苷含量,測定結果見表10。
表10 HPLC法和UV法測定西紅花中色-香-味的含量
從表10可看出,國產不同來源的28批西紅花色香味三類成分的含量有較大差異。苦藏花素含量最高的(S27)是含量最低的(S7)的近3倍。紫外測定的總西紅花苷含量最高的(S27)是含量最低的(S26)的1.5倍。而藏花醛含量最高的(S7)是含量最低的(S19)的25倍。藏花醛在新鮮西紅花柱頭中基本不存在,是新鮮西紅花在加熱干燥過程中由苦藏花素水解后生成苷元,在高溫作用下進一步脫水生成。即藏花醛實際上不是新鮮西紅花的原始積累產物,是一個采后“干燥脅迫”誘導產物。后期研究表明,該成分的含量與干燥溫度有關,S7苦藏花素含量最低,可能是干燥時溫度高,苦藏花素熱降解,轉化成藏花醛。
3.基于TOPSIS分析樣品的質量優劣
西紅花藥材質量與色-香-味多組分的含量有關,是一個典型的多指標決策問題。由于各指標成分含量不同,且在數量級上也存在較大差異,簡單加和顯然不能客觀全面反映樣品質量,因此需要選擇合理的綜合評價方法,以兼顧各種成分在質量評價中所占權重均等。TOPSIS分析法是一種有效的多指標較理想的綜合評價方法,其中心思想是首先確定各項指標的正理想值A+和負理想值A-,即各指標的最優解和最劣解。然后求出各樣品與正、負理想值之間的加權歐式距離D+和D-,從而求出各樣品與理想方案的貼近程度Ci,作為評價方法優劣的標準。即靠即近正理想和遠離負理想的程度,來對方案進行排序,相對貼近度越大,說明越靠近正理想而遠離負理想,方案越優;反之,方案越劣。
采用DPS數據處理系統,以28批樣品中的7個指標成分組成28×11矩陣,基于TOPSIS通過Matlab軟件編程實現其貼近度大小,利用貼近度(Ci)對28批樣品進行綜合排序,結果見表11。首先分別找出7個指標的正負理想如下:
正理想(A+):19.84,18.98,8.84,27.60,28.90,4.06,0.999
負理想(A-):6.65,11.59,4.25,16.89,19.05,0.16,0.934
表11 TOPSIS綜合評價結果
TOPSIS評價結果顯示,前5批由優至劣順次排序的樣品為:S27(浙江杭州),S1(浙江建德三都西紅花合作社),S15(浙江余杭),S22(江西南昌),S2(浙江建德三都西紅花合作社)。28批樣品排序后5批的樣品為:S11(浙江湖州安吉),S25(西藏),S26(浙江湖州),S9(浙江金華浦江縣),S5(浙江建德楊樹橋鎮)。本研究基于“色-香-味多指標定量結合指紋圖譜相似度,應用TOPSIS法評價”,可全面表征不同來源西紅花藥材的化學成分的差別,比各項指標簡單加和更能客觀全面反映西紅花藥材質量,可以為西紅花藥材產地選擇,臨床應用及質量快速評價提供參考。在此基礎上,若能將藥效、毒性與物質基礎結合起來,深入開展譜效學研究,則更有利于闡明中藥作用機制,完善西紅花的質量評價體系,為用藥安全提供進一步保障。
從TOPSIS評價結果可看出,S5的貼近度(Ci)最小,質量最差。從表10數據可看出,該樣品的苦藏花素,西紅花苷類成分的含量均比S7高,指紋圖譜的相似度也比S7高,但藏花醛含量比S7低,如果從色香味結果的表面去分析,S7質量相對最差。
2015年版《中國藥典》用HPLC法測定西紅花苷-Ⅰ和西紅花苷-Ⅱ的含量,規定兩者總含量大于10%為合格品,28批樣品均為合格品,如按照中國藥典評價,質量最差的是S26。因此,僅由西紅花苷-Ⅰ和西紅花苷-Ⅱ總量評價西紅花有一定局限性。
因此,本研究基于“色-香-味多指標定量結合指紋圖譜相似度,應用TOPSIS法評價”,所得出的樣品質量優劣順序較簡單加和法將更能綜合反映各樣品的綜合質量。
實例5:色-香-味多指標定量結合指紋圖譜對進口西紅花的質量評價
國產西紅花色香味多組分指紋圖譜在進口西紅花鑒別中的應用,結果已表明,12批進口西紅花的相似度均在0.9以下,均比國產西紅花的相似度低,為了進一步考察本發明建立的“色-香-味多指標定量結合指紋圖譜相似度綜合評價進口西紅花的質量”,將在前面研究基礎上,利用TOPSIS法評價國產與進口西紅花的質量差異。色香味含量和相似度結果見表12。
表12 HPLC法和UV法測定進口西紅花中色-香-味的含量
比較表10和表11,可看出進口西紅花其西紅花苷類和苦藏花素的含量均比國產西紅花低,但藏花醛的含量進口西紅花整體比國產西紅花高。進一步利用TOPSIS綜合評價,通過比較貼近度(Ci),從排序結果發現,排在后十二位的除國產S5號外,都是進口西紅花,說明進口西紅花距離正理想遠而距離負理想近;而且,負理想中的數據基本來自于進口西紅花組,正理想中的數據基本來自國產西紅花組。
因此,TOPSIS綜合評價結果表明,國產西紅花質量整體優于進口西紅花。可能與國內外西紅花栽培方式和采后加工等因素有關。種球大小與西紅花苷和苦藏花素含量有關,種球越大,積累西紅花苷和苦藏花素含量越高。進口西紅花采用連續栽培方式,種球較小,成分含量較低。而國產西紅花采用二段式栽培方式,室內開花,開花后,種球再種于室外,進行營養生長,培育種球,種球較大,成分含量較高。
上述說明示出并描述了本發明的若干優選實施例,但如前所述,應當理解本發明并非局限于本文所披露的形式,不應看作是對其他實施例的排除,而可用于各種其他組合、修改和環境,并能夠在本文所述發明構想范圍內,通過上述教導或相關領域的技術或知識進行改動。而本領域人員所進行的改動和變化不脫離本發明的精神和范圍,則都應在本發明所附權利要求的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!