一種改進的5?胺甲基尿嘧啶修飾泡塑富集金的分析方法與流程
本發明屬于巖石礦物元素分析技術領域,涉及一種測定地質樣品中金的分析方法,尤其涉及一種應用5-胺甲基尿嘧啶修飾泡塑富集地質樣品中金的分析方法。
背景技術:
黃金,稀有而珍貴,并且耐腐蝕,自古以來被視為五金之首。除了做為各國重要的戰略儲備資源,在工業與科學技術上也有重要用途。如:可以應用于儀器儀表制造業,作為精密電位計關鍵材料;廣泛用于電子設備、半導體器材和微型電路中做導體材料。另外,在冶金、航海、航空、航天、電子器械、石油化工等領域也有重要應用。我國為貴金屬資源比較貧乏的國家,研究金的測定分析方法具有非常重要的意義,有利于化探樣品找礦和礦產資源的可持續發展。
金在地殼中的平均含量為1×10-9,在礦石中以自然金、固溶體類金礦物、化合物類金礦物存在,并伴有“塊金效應”。因此,一般采用比較大的取樣量克服“塊金效應”,由于含量低,分析前需要經過分離富集,以減少干擾,提高分離靈敏度。礦石中金的測定方法有經典的火試金法、共沉淀、活性炭富集和泡塑富集等。采用火試金和硫鎳試金分析,配料復雜,耗時長,手續繁瑣且空白值高;采用共沉淀流程長,分析樣品量有限,無法克服“塊金效應”。目前應用較多的是活性炭和泡塑。但是活性炭的選擇性非常差,富集過程中,干擾離子也同時被吸附在活性炭上,影響分析準確度。泡塑吸附法簡便易行,載入合適的載體可以提高金的吸附率,增強泡塑的抗干擾能力,此方法已經有較多的研究。但是泡塑吸附金的缺點是吸附容量偏低。本方法采用5-胺甲基尿嘧啶做為修飾配體,以二醛為連接臂,在水相中合成5-胺甲基尿嘧啶修飾泡塑。5-胺甲基尿嘧啶和氨基泡塑分別和二醛生產亞胺,將5-胺甲基尿嘧啶以共價鍵的形式修飾到泡塑表面。該方法由于是在水相中合成,方法更加綠色,環保。并且5-胺甲基尿嘧啶修飾泡塑的吸附容量提高至100mg/g。
技術實現要素:
為解決泡塑富集金時,常規泡塑吸附容量偏低的問題,本發明提供一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法,其按照先后順序包括以下步驟:
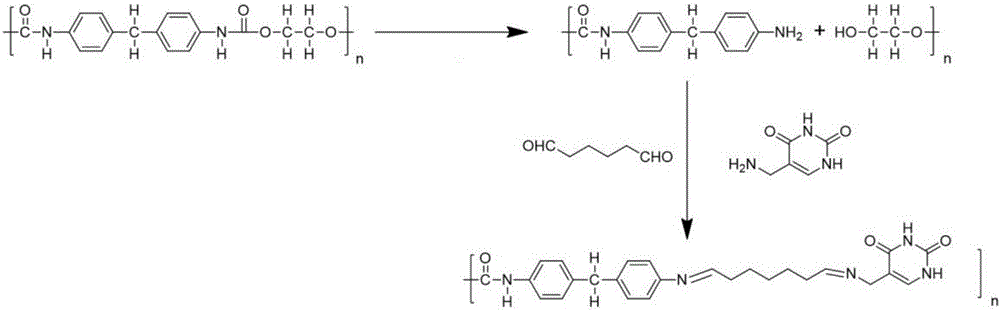
(1)首先制作5-胺甲基尿嘧啶修飾的泡塑:將泡塑用粉碎機打碎,過篩。過篩后的泡塑粒徑保持在100~200μm之間。然后,在2mol/L鹽酸中加熱煮沸,水解2小時,用蒸餾水洗至中性;在真空干燥箱中40℃,干燥8小時。即得到氨基泡塑。稱取1.0~2.0克氨基泡塑到250毫升圓底燒瓶中,加入100毫升水,再加入2~3毫升二醛溶液,常溫下避光攪拌1~2個小時,接著再加入0.05~0.2克5-胺甲基尿嘧啶,繼續反應8~12小時。合成出5-胺甲基尿嘧啶修飾的泡塑,其機理如圖2所示。
(2)吸附柱的制作:將0.25克5-胺甲基尿嘧啶修飾的泡塑裝入吸附柱,吸附柱進出口都裝上篩板,吸附柱規格為12毫升;該吸附柱使用前,先分別用20毫升0.1mol/L鹽酸和20毫升去離子水淋洗;
(3)準確稱取10.0克地質樣品于瓷坩堝中,將瓷坩堝置于低溫馬弗爐中,升溫到650~700℃灼燒1小時,取出冷卻,將樣品倒入200毫升帶螺紋蓋的特氟龍燒杯中,加10mL水潤濕,再加入40毫升50%王水,和5毫升HF,攪勻,打開蓋,置于電熱板上加熱溶解至近干。反復2次。用稀鹽酸定容至100毫升,并用氨水調節溶液的pH=0.1~2.0;
(4)將第三步驟中所得溶液,取25毫升上清液,使該溶液以1.0~4.0mL min-1的流速通過吸附柱,對金進行分離富集;吸附柱下方連接蠕動泵,通過蠕動泵來控制流速(如圖5所示);
(5)采用5毫升5g/L硫脲——1%(V/V)鹽酸溶液淋洗吸附柱,保持流速在1.0~2.0mL min-1。洗脫后的洗脫液,采用電感耦合等離子光譜,在波長金(242.80)處進行測定。
優選的是,步驟(1)中,所述將泡塑需要在2mol/L鹽酸中加熱煮沸,水解2小時,以便使泡塑表面形成氨基。得到氨基泡塑后。需要與二醛先在常溫下避光攪拌1~2個小時,氨基泡塑表面的氨基和二醛中的一個醛基反應,形成亞胺鍵,使二醛連接在氨基泡塑表面。接著再加入5-胺甲基尿嘧啶,繼續反應8~12小時。合成出5-胺甲基尿嘧啶修飾的泡塑。本發明經過大量實驗證明,通過上述步驟,5-胺甲基尿嘧啶可以牢固的修飾在泡塑表面,有利于對金的吸附作用。
在上述任一方案中優選的是,步驟(2)中,所述0.25克5-胺甲基尿嘧啶修飾的泡塑裝入吸附柱,吸附柱進出口都裝上篩板,吸附柱規格為12毫升;該吸附柱使用前,先分別用20毫升0.1mol/L鹽酸和20毫升去離子水淋洗,洗去雜質。本發明經過大量實驗證明,5-胺甲基尿嘧啶修飾的泡塑在這一用量條件下,可以定量富集,保留地質樣品中的金,測定結果較準確。
在上述任一方案中優選的是,步驟(4)中,所述取25毫升上清液,使該溶液以1.0~4.0mL min-1的流速通過吸附柱,對金進行分離富集;吸附柱下方連接蠕動泵,通過蠕動泵來控制流速(如圖5所示)。本發明經過大量實驗證明,需要取25毫升上清液做吸附實驗,該溶液在1.0~4.0mL min-1的流速下通過吸附柱,金可以定量保留在吸附劑上,測定結果較準確。
在上述任一方案中優選的是,步驟(5)中,所述采用5毫升5g/L硫脲——1%(V/V)鹽酸溶液淋洗吸附柱,保持流速在1.0~2.0mL min-1。本發明經過大量實驗證明,采用至少5毫升5g/L硫脲——1%(V/V)鹽酸洗脫液可以定量將金從吸附劑上洗脫下來,并且洗脫劑流速應該控制在1.0~2.0mL min-1,測定結果更準確。
本發明提供一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法。5-胺甲基尿嘧啶修飾泡塑對金的選擇性好,吸附容量高。整個實驗過程,分析結果準確,可靠。
附圖說明
圖1為按照本發明的利用改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的分析流程圖;
圖2為按照本發明的利用改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的合成5-胺甲基尿嘧啶修飾泡塑的合成路線圖。
圖3為按照本發明的利用改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的吸附容量實驗圖。從圖3可以看出,5-胺甲基尿嘧啶修飾泡塑吸附容量在100mg/g,遠高于普通泡塑(15mg/g)。
圖4為按照本發明的利用改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的泡塑和5-胺甲基尿嘧啶修飾泡塑的紅外光譜圖。通過紅外光譜的表征。氮氫鍵的伸縮振動,由泡塑的3299.1cm-1移動到5-胺甲基尿嘧啶修飾泡塑的3409.4cm-1。并且5-胺甲基尿嘧啶修飾泡塑的峰面積有了顯著增加,其增加的原因主要是由于5-胺甲基尿嘧啶配體中含有的二級胺N-H鍵。5-胺甲基尿嘧啶修飾泡塑出現了兩個額外的峰1692.0cm-1和1732.0cm-1。1692.0cm-1峰的形成主要是由于反應過程中形成了亞胺鍵,1732.0cm-1峰的形成主要是由于5-胺甲基尿嘧啶含有較多C=O鍵。1448.0cm-1峰的形成主要是5-胺甲基尿嘧啶配體中含有C-H鍵。
圖5為按照本發明的利用改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的固相萃取流速控制圖。
具體實施方式
為了更進一步了解本發明的發明內容,下面將結合具體實施例詳細闡述本發明。
實施例一:
如圖1所示,一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法,其按照先后順序包括以下步驟:
(1)首先制作5-胺甲基尿嘧啶修飾的泡塑:將泡塑用粉碎機打碎,過篩。過篩后的泡塑粒徑保持在100~200μm之間。然后,在2mol/L鹽酸中加熱煮沸,水解2小時,用蒸餾水洗至中性;在真空干燥箱中40℃,干燥8小時。即得到氨基泡塑。稱取1.0克氨基泡塑到250毫升圓底燒瓶中,加入100毫升水,再加入2毫升己二醛溶液,常溫下避光攪拌2個小時,接著再加入0.05克5-胺甲基尿嘧啶,繼續反應8小時。合成出5-胺甲基尿嘧啶修飾的泡塑。
(2)吸附柱的制作:將0.25克5-胺甲基尿嘧啶修飾的泡塑裝入吸附柱,吸附柱進出口都裝上篩板,吸附柱規格為12毫升;該吸附柱使用前,先分別用20毫升0.1mol/L鹽酸和20毫升去離子水淋洗;
(3)準確稱取10.0克國家標準物質GBW070012于瓷坩堝中,將瓷坩堝置于低溫馬弗爐中,升溫到650~700℃灼燒1小時,取出冷卻,將樣品倒入200毫升帶螺紋蓋的特氟龍燒杯中,加10mL水潤濕,再加入40毫升50%王水,和5毫升HF,攪勻,打開蓋,置于電熱板上加熱溶解至近干。反復2次。用稀鹽酸定容至100毫升,并用氨水調節溶液的pH=0.1~2.0;
(4)將第三步驟中所得溶液,取25毫升上清液,使該溶液以1.0mL min-1的流速通過吸附柱,對金進行分離富集;吸附柱下方連接蠕動泵,通過蠕動泵來控制流速(如圖5所示);
(5)采用5毫升5g/L硫脲——1%(V/V)鹽酸溶液淋洗吸附柱,保持流速在1.0mL min-1。洗脫后的洗脫液,采用電感耦合等離子光譜,在波長金(242.80)處進行測定。
步驟(5)中,選取國家標準物質GBW070012對上述測定方法進行可行性評價。利用上述測定方法對國家標準物質GBW070012中金的測定值與標準值的對比如表一所示:
表一 國家標準物質GBW070012中金的測定值與標準推薦值的對比(微克/克),測量次數為3次。
注:Mean代表平均值;1s代表一倍的標準偏差。
從表一中的實驗數據可知,上述一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的準確度和精密度滿足科研工作與野外勘察的要求,具有良好的可行性。
實施例二:
如圖1所示,一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法,其按照先后順序包括以下步驟:
(1)首先制作5-胺甲基尿嘧啶修飾的泡塑:將泡塑用粉碎機打碎,過篩。過篩后的泡塑粒徑保持在100~200μm之間。然后,在2mol/L鹽酸中加熱煮沸,水解2小時,用蒸餾水洗至中性;在真空干燥箱中40℃,干燥8小時。即得到氨基泡塑。稱取1.5克氨基泡塑到250毫升圓底燒瓶中,加入100毫升水,再加入3毫升庚二醛溶液,常溫下避光攪拌2個小時,接著再加入0.10克5-胺甲基尿嘧啶,繼續反應8小時。合成出5-胺甲基尿嘧啶修飾的泡塑。
(2)吸附柱的制作:將0.25克5-胺甲基尿嘧啶修飾的泡塑裝入吸附柱,吸附柱進出口都裝上篩板,吸附柱規格為12毫升;該吸附柱使用前,先分別用20毫升0.1mol/L鹽酸和20毫升去離子水淋洗;
(3)準確稱取10.0克國家標準物質GBW07807于瓷坩堝中,將瓷坩堝置于低溫馬弗爐中,升溫到650~700℃灼燒1小時,取出冷卻,將樣品倒入200毫升帶螺紋蓋的特氟龍燒杯中,加10mL水潤濕,再加入40毫升50%王水,和5毫升HF,攪勻,打開蓋,置于電熱板上加熱溶解至近干。反復2次。用稀鹽酸定容至100毫升,并用氨水調節溶液的pH=0.1~2.0;
(4)將第三步驟中所得溶液,取25毫升上清液,使該溶液以1.0mL min-1的流速通過吸附柱,對金進行分離富集;吸附柱下方連接蠕動泵,通過蠕動泵來控制流速(如圖5所示);
(5)采用5毫升5g/L硫脲——1%(V/V)鹽酸溶液淋洗吸附柱,保持流速在1.0mL min-1。洗脫后的洗脫液,采用電感耦合等離子光譜,在波長金(242.80)處進行測定。
步驟(5)中,選取國家標準物質GBW07807對上述測定方法進行可行性評價。利用上述測定方法對國家標準物質GBW07807中金的測定值與標準值的對比如表一所示:
表二 國家標準物質GBW07807中金的測定值與標準推薦值的對比(微克/克),測量次數為3次。
注:Mean代表平均值;1s代表一倍的標準偏差。
從表二中的實驗數據可知,上述一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的準確度和精密度滿足科研工作與野外勘察的要求,具有良好的可行性。
實施例三:
如圖1所示,一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法,其按照先后順序包括以下步驟:
(1)首先制作5-胺甲基尿嘧啶修飾的泡塑:將泡塑用粉碎機打碎,過篩。過篩后的泡塑粒徑保持在100~200μm之間。然后,在2mol/L鹽酸中加熱煮沸,水解2小時,用蒸餾水洗至中性;在真空干燥箱中40℃,干燥8小時。即得到氨基泡塑。稱取2克氨基泡塑到250毫升圓底燒瓶中,加入100毫升水,再加入3毫升壬二醛溶液,常溫下避光攪拌2個小時,接著再加入0.15克5-胺甲基尿嘧啶,繼續反應8小時。合成出5-胺甲基尿嘧啶修飾的泡塑。
(2)吸附柱的制作:將0.25克5-胺甲基尿嘧啶修飾的泡塑裝入吸附柱,吸附柱進出口都裝上篩板,吸附柱規格為12毫升;該吸附柱使用前,先分別用20毫升0.1mol/L鹽酸和20毫升去離子水淋洗;
(3)準確稱取10.0克國家標準物質GBW07809于瓷坩堝中,將瓷坩堝置于低溫馬弗爐中,升溫到650~700℃灼燒1小時,取出冷卻,將樣品倒入200毫升帶螺紋蓋的特氟龍燒杯中,加10mL水潤濕,再加入40毫升50%王水,和5毫升HF,攪勻,打開蓋,置于電熱板上加熱溶解至近干。反復2次。用稀鹽酸定容至100毫升,并用氨水調節溶液的pH=0.1~2.0;
(4)將第三步驟中所得溶液,取25毫升上清液,使該溶液以1.0mL min-1的流速通過吸附柱,對金進行分離富集;吸附柱下方連接蠕動泵,通過蠕動泵來控制流速(如圖5所示);
(5)采用5毫升5g/L硫脲——1%(V/V)鹽酸溶液淋洗吸附柱,保持流速在1.0mL min-1。洗脫后的洗脫液,采用電感耦合等離子光譜,在波長金(242.80)處進行測定。
步驟(5)中,選取國家標準物質GBW07809對上述測定方法進行可行性評價。利用上述測定方法對國家標準物質GBW07809中金的測定值與標準值的對比如表一所示:
表三 國家標準物質GBW07809中金的測定值與標準推薦值的對比(微克/克),測量次數為3次。
注:Mean代表平均值;1s代表一倍的標準偏差。
從表三中的實驗數據可知,上述一種改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法的準確度和精密度滿足科研工作與野外勘察的要求,具有良好的可行性。
ICP光譜型號為IRIS Advantage ICP-OES,生產廠家為美國熱電公司。儀器條件為::ICP射頻功率1150W,輔助氣流量0.5L/min,霧化氣壓力27PSI,冷卻氣流量15.0L/min,曝光時間:短波15秒;長波8秒。
本領域技術人員不難理解,本發明的利用改進的5-胺甲基尿嘧啶修飾泡塑富集金的分析方法,包括上述本發明說明書的發明內容和具體實施方式部分以及附圖所示出的各部分的任意組合,限于篇幅并為使說明書簡明而沒有將這些組合構成的各方案一一描述。凡在本發明的精神和原則之內,所做的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!