一種具有極高選擇性的測定鋅電解液中Co2+含量的絡合物吸附波極譜法的制作方法
本發明涉及一種直接檢測鋅電解液中微量或痕量Co2+的絡合物吸附波極譜法。
背景技術:
鋅是支撐國民經濟和國防軍工發展的基礎原料和戰略性物資,目前80%左右的鋅是通過濕法冶金產生的。濕法煉鋅工藝依次主要包括磨礦、浸出、凈化和電解過程。其中凈化工序是用來控制電解液中雜質離子濃度的關鍵工序,通過待凈化液中雜質的種類和含量來判斷需要加入的除雜試劑的種類和用量。目前大部分冶煉廠仍采用手工間歇性的傳統方法來分析有關雜質的種類和含量,這些方法過程繁瑣、耗時,導致檢測信息反饋滯后時間長。這使得凈化工序的工藝參數無法實現實時優化調整,往往須采用過量加入鋅粉以保證除雜效果,導致凈化后料液雜質離子濃度波動大、鋅粉大量浪費、生產成本高等問題。因此,開發檢測步驟簡單、速度快,適用于在線檢測的方法至關重要。在電解過程中,雜質金屬離子在陰極放電析出嚴重地影響析出鋅的結晶狀態、電積過程的電流效率和電解鋅的質量。鈷是濕法煉鋅浸出液中最難除去的雜質之一,且對鋅的電沉積危害非常大,當其濃度超過一定值時,就會引起“燒板”現象和降低電流效率等危害,因此需要嚴格監控鋅電解液中鈷的含量。
微量Co2+的測定方法很多,主要有分光光度法,原子吸收法,原子發射光譜法,極譜法,色譜法,質譜法等。由于鋅電解液中Zn2+的含量很高(130~170g/L),鋅鈷的質量比最高可達到107倍以上,以至嚴重干擾鋅電解液中Co2+的測定,這些方法都不能滿足簡便快速實現鈷測定的要求。目前部分冶煉廠采用亞硝基R鹽吸光光度法來測定Co2+,但方法靈敏度低,線性范圍較窄(0.3~5μg·mL-1),且須反復多次加熱煮沸,分析速度慢,難以快速準確地得到分析結果,不能在線監測除雜效果和凈化液質量。所以尋找新的測試方法至關重要。
絡合物吸附波極譜法一般靈敏度高,選擇性好,儀器簡單,分析速度快,非常適合微量或痕量元素的測定和在線分析檢測儀器的研究開發,目前關于絡合物吸附波測定水溶液中的Co2+已有報道,而且大部分方法采用中性或堿性(多為氨性)介質,Co2+的峰電位都在-1.0V(vs.SCE)左右,與Zn2+的峰電位很接近,因而受Zn2+的干擾相當嚴重,當用來測定鋅電解液中含量較低的鈷時Co2+波則完全被Zn2+波掩蓋而無法測定,若采用預處理方法沉淀Zn2+,不僅操作麻煩,而且在在線分析中,沉淀的生成容易堵塞管路,不易清理。
技術實現要素:
本發明的目的是針對鋅電解液中Co2+的測定,提供一種簡單,快捷,高選擇性和高靈敏度的能夠用于在線分析的絡合物吸附波極譜檢測方法。
本發明通過以下方案來實現:
一種具有極高選擇性的測定鋅電解液中Co2+含量的絡合物吸附波極譜法,包括將待測樣品鋅電解液與檢測體系反應,測定反應混合物中產生的高選擇性和靈敏度的Co2+絡合物吸附極譜波,獲得二階導數波峰電流,從而計算待測樣品鋅電解液中的Co2+濃度;所述檢測體系包括底液氨-氯化銨緩沖液,掩蔽劑乙二胺四乙酸鹽,絡合劑丁二酮肟或者絡合劑丁二酮肟和亞硝酸鈉。
本發明方法檢測體系選取乙二胺四乙酸鹽作為鋅電解液中基體成分Zn2+和其他金屬離子的掩蔽劑,丁二酮肟、或丁二酮肟和亞硝酸鈉作為待測離子Co2+的絡合劑(或混配絡合劑),其中丁二酮肟作為測Co2+的主絡合劑、亞硝酸鈉作為輔助絡合增敏劑,以氨-氯化銨緩沖液為底液(介質),將待測樣品鋅電解液與檢測體系反應,測定反應混合物中產生的高選擇性和靈敏度的Co2+絡合物吸附極譜波,鋅和電解液中其它離子對Co2+檢測都不產生干擾。在常溫下測定鋅電解液中微量或痕量Co2+時,該方法不需預處理可直接用示波極譜儀等儀器測定。本發明不僅可以完全掩蔽Zn2+,消除電解液中高濃度基體成分Zn2+(鋅鈷質量比≥107)對Co2+測定的影響,而且可完全掩蔽鋅電解液中其他共存雜志離子的干擾,是定性和定量測定鋅電解液中微量或痕量鈷的高選擇性和靈敏的方法。
優選地,所述反應混合物中乙二胺四乙酸鹽的摩爾濃度與鋅電解液中Zn2+摩爾濃度的比值為1~5,進一步優選為1.1。
優選地,所述乙二胺四乙酸鹽為乙二胺四乙酸二鈉,具體可用EDTA的氫氧化鈉溶液,所述反應混合物中EDTA的濃度為0.005~0.007mol/L,優選為0.006mol/L。
優選地,所述EDTA的氫氧化鈉溶液中EDTA的濃度為0.25~0.35mol/L,進一步優選為0.30mol/L。
優選地,所述反應混合物中氨-氯化銨的摩爾濃度為0.3~0.6mol/L,優選為0.48mol/L。
優選地,所述氨-氯化銨緩沖液的濃度為2.0-4.0mol/L,優選為3.0mol/L,其中NH3與NH4Cl的摩爾比為1:9。
當待測樣品鋅電解液中Co2+濃度為1.0×10-10~4.0×10-4mol/L,優選為1.2×10-9~4.0×10-4mol/L時,所述絡合劑為丁二酮肟.
當待測樣品鋅電解液中Co2+濃度為2.0×10-12~3.2×10-6mol/L,優選為5.0×10-11~3.2×10-6mol/L時;所述絡合劑為丁二酮肟和亞硝酸鈉。
優選地,所述反應混合物中丁二酮肟的摩爾濃度為7.6×10-4~3.8×10-3mol/L,優選為2.7×10-3mol/L;所述反應混合物中亞硝酸鈉的摩爾濃度為0.16~0.48mol/L,優選為0.32mol/L。
優選地,所述丁二酮肟以丁二酮肟的乙醇溶液的形式添加,其濃度為5~12g/L,進一步優選為11g/L。
優選地,所述亞硝酸鈉以亞硝酸鈉溶液的形式添加,其濃度為2.0~6.0mol/L,進一步優選為4.0mol/L。
本發明方法不需預處理可直接可采用示波極譜儀等儀器進行測定,例如采用JP-303E型示波極譜儀。
測定條件包括:
起始電壓:-100~-1050mV,優選為-900mV;
終止電壓:-1250~-1500mV,優選為-1300mV;
掃描速率:100~500mV/s,優選為300mV/s;
靜止時間:5~14s,優選為6s;
滴汞周期:5~18s,優選為9s。
當電解新液、電解廢液、除鈷后液等待測樣品鋅電解液中Co2+濃度較低(例如痕量)時,所述絡合劑為丁二酮肟和亞硝酸,即采用丁二酮肟-EDTA-亞硝酸鈉-“氨-氯化銨緩沖液”體系(簡稱體系一)測定Co2+,Co2+線性范圍為:5.0×10-11~3.2×10-6mol/L(見圖1),相關系數:0.9997,檢出限:2.0×10-12mol/L。具體地,所述痕量指鋅電解液中Co2+濃度為2.0×10-12~3.2×10-6mol/L,優選為5.0×10-11~3.2×10-6mol/L。
當待測樣品鋅電解液中Co2+濃度較高(例如微量)時,可適當稀釋后再采用上述“體系一”進行測定。
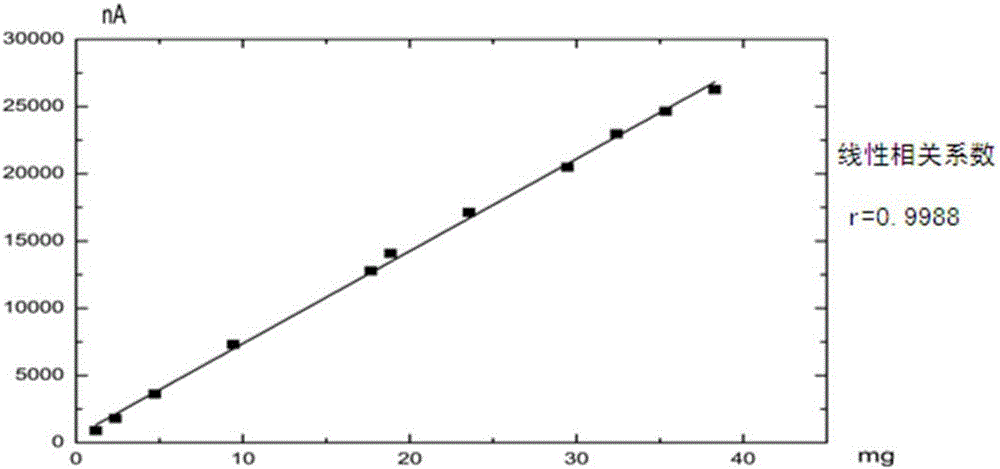
為避免反復稀釋的麻煩,當中性上清液等待測樣品鋅電解液中Co2+濃度較高(例如微量)時,所述絡合劑為丁二酮肟,即采用丁二酮肟-EDTA-“氨-氯化銨緩沖液”體系(簡稱體系二)測定Co2+,即省略加入亞硝酸鈉的步驟;Co2+線性范圍為:1.2×10-9~4.0×10-4mol/L(見圖2),相關系數:0.9988,檢出限:1.0×10-10mol/L。具體地,所述微量指鋅電解液中Co2+濃度為1.0×10-10~4.0×10-4mol/L,優選為1.2×10-9~4.0×10-4mol/L。
具體地,上述測定方法包括以下步驟:
1)將一定量的待測樣品鋅電解液與一定量的所述檢測體系反應,測定反應混合物中產生的Co2+絡合物吸附極譜波(采用示波極譜儀攝圖,下同),獲得二階導數波峰電流I1;
2)分別取與步驟1)等量的待測樣品鋅電解液、與步驟1)等量的所述檢測體系及一定量的濃度為a mol/L的Co2+標準溶液,將三者反應測定反應混合物中產生的Co2+絡合物吸附極譜波,獲得二階導數波峰電流I2;
3)按下式計算待測樣品鋅電解液中的Co2+濃度,單位mol/L:
a I1/(I2–I1);
優選地,a為5.0×10-11-4.0×10-4。
更具體地,上述測定方法包括以下步驟:
1)量取0.10mL的待測樣品鋅電解液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,或者進一步再加入亞硝酸鈉溶液4.0mL,靜止5min,把所得混合溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波(采用示波極譜儀攝圖,下同),獲得二階導數波峰電流I1;
2)量取0.10mL濃度為a mol/L的Co2+標準溶液和0.10mL的待測樣品鋅電解液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,或者進一步再加入亞硝酸鈉溶液4.0mL,靜止5min,把所得混合溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波(采用示波極譜儀攝圖,下同),獲得二階導數波峰電流I2;
3)按下式計算待測樣品鋅電解液中的Co2+濃度,單位mol/L:
a I1/(I2–I1);
其中,
丁二酮肟的乙醇溶液濃度為11g/L;
EDTA的氫氧化鈉溶液中EDTA的濃度為0.30mol/L;
氨-氯化銨緩沖液的濃度為3.0mol/L,其中NH3與NH4Cl的摩爾比為1:9;
亞硝酸鈉溶液濃度為4.0mol/L;
a為5.0×10-11-4.0×10-4;
當所述待測樣品鋅電解液中Co2+濃度為2.0×10-12~3.2×10-6mol/L,優選為5.0×10-11~3.2×10-6mol/L時;所述絡合劑為丁二酮肟和亞硝酸鈉;
當待測樣品鋅電解液中Co2+濃度為1.0×10-10~4.0×10-4mol/L,優選為1.2×10-9~4.0×10-4mol/L時,所述絡合劑為丁二酮肟。
步驟1)和步驟2)測定Co2+絡合物吸附極譜波的條件相同,具體包括:
儀器:JP-303E型示波極譜儀;
起始電壓:-900mV;
終止電壓:-1300mV;
掃描速率:300mV/s;
靜止時間:6s;
滴汞周期:9s。
當所述待測樣品鋅電解液中含有微量的Co2+,優選Co2+濃度≥3.2×10-6mol/L時,可將待測樣品鋅電解液適當稀釋后再進行測定,例如稀釋10-1000倍。
本發明所述待測樣品包括鋅電解液、鋅電解新液、中性上清液等。
上述兩體系中對Co2+測定相對誤差不超過5%。下列質量倍數的離子:Zn2+(4×107)、W(VI)和Mn2+(3×104)、Mo(VI)和Hg2+(1×104)、Pb2+(3×103)、Cu2+和Cd2+(1000)、Sb3+(500)、Ni2+(450)、As3+(110)、Fe3+(20)及常見陰離子均不干擾測定。
用上述兩個體系直接測定鋅電解液中鈷,鋅電解液中其它共存離子對Co2+的測定均無干擾,滿足測定要求。
本發明的思路是要求分析方法操作簡單,選擇性高,靈敏度高,分析時間短,不需要對樣品進行預處理,儀器及試劑價格便宜,不產生沉淀,能夠用于在線分析。氨羧絡合劑具有廣泛而強大的絡合能力,與多種金屬離子形成的絡合物很穩定,且能溶于水。乙二胺四乙酸鹽(EDTA)作為應用最廣泛的氨羧絡合劑,性質穩定,價格便宜,能與大部分金屬離子形成配位比為1:1的穩定螯合物。實驗發現,Co2+與丁二酮肟形成的絡合物的穩定常數非常大,甚至遠超過Co2+與EDTA形成絡合物的穩定常數,而其它金屬離子與丁二酮肟生成的絡合物其穩定性則小的多。在丁二酮肟存在下,EDTA與鋅電解液中除Co2+之外所有金屬離子發生螯合反應,且在測定鈷的條件下其螯合物不產生絡合物吸附波,因而所有與Co2+共存的金屬離子都被掩蔽,而Co2+與丁二酮肟(或丁二酮肟及亞硝酸鈉)形成的絡合物在氨-氯化銨底液中則產生靈敏的絡合物吸附波。因此,此方法對于鋅電解液中Co2+的測定具有非常好的選擇性和靈敏度。
本發明采用EDTA作為基體成分鋅和共存雜質金屬離子的掩蔽劑,為了避免因測定較高含量鈷須聯結稀釋裝置使在線檢測儀器結構復雜化,可采用兩個體系分別對微量和痕量Co2+進行測定。在痕量時采用丁二酮肟-EDTA-亞硝酸鈉-氨緩沖體系測定Co2+,在微量時采用丁二酮肟-EDTA-氨緩沖體系測定Co2+,試劑簡單方便配制,大大提高了自動化程度。本發明方法不僅可以完全掩蔽Zn2+,消除Zn2+波對Co2+測定的影響,而且也可掩蔽鋅電解液中其他共存雜質金屬離子的干擾,選擇性極好,不需要對鋅電解液預處理,沒有沉淀生成,分析速度快,適合在線分析檢測使用。
本發明與現有方法相比,具有如下有益效果:
1.無需對被測鋅電解液進行預處理;
2.線性范圍寬,對整個工藝流程的Co2+都可以檢測;
3.所用儀器價格便宜,操作簡單。所用試劑種類少,穩定性好,價格便宜;
4.分析速度快;
5.無沉淀生成;不僅適用于實驗室中常規分析,更易實現自動化,便于在線檢測中使用。
附圖說明
圖1為實施例1痕量(較低濃度)鈷測定體系的線性回歸曲線;
圖2為實施例2微量(較高濃度)鈷測定體系的線性回歸曲線;
圖3為實施例3(采用體系一)測定鋅電解新液中Co2+的二階導數極譜波;
圖4為實施例4(采用體系二)測定中性上清液中Co2+的二階導數極譜波;
圖5為實施例5(采用體系一)測定中性上清液中Co2+的二階導數極譜波。
具體實施方式
以下實施例用于說明本發明,但不用來限制本發明的范圍。實施例中未注明具體技術或條件者,按照本領域內的文獻所描述的技術或條件,或者按照產品說明書進行。所用試劑或儀器未注明生產廠商者,均為可通過正規渠道商購買得到的常規產品。
以下實施例所用試劑及規格:
丁二酮肟的乙醇溶液:11g/L;EDTA的氫氧化鈉溶液中EDTA的濃度:0.30mol/L;氨-氯化銨緩沖液:3.0mol/L;NH3與NH4Cl的摩爾比為1:9;亞硝酸鈉溶液:4.0mol/L。
以下實施例測試條件:
儀器:JP-303E型示波極譜儀;起始電壓:-900mV;終止電壓:-1300mV;掃描速率:300mV/s;靜止時間:6s;滴汞周期:9s。
以下實施例待測樣品鋅電解液、鋅電解新液、中性上清液由湖南省株洲冶煉廠提供。
實施例1 痕量(較低濃度)鈷測定體系的線性回歸曲線的制作方法
(1)取Co2+標液(標準溶液)用二次蒸餾水稀釋成適宜濃度的Co2+。
(2)分別量取2.5×10-7、5.0×10-7、1.0×10-6、2.0×10-6、4.0×10-6、8.0×10-6和1.6×10-5mol/L的Co2+標液各0.10mL于7個50mL容量瓶中,分別向容量瓶中加入2.6mol/L Zn2+0.10mL,丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,亞硝酸鈉溶液4.0mL于11個50mL容量瓶中,靜止5min,分別把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得測得各濃度Co2+對應的二階導數波峰電流,繪制線性回歸曲線如圖1所示(橫坐標表示加入容量瓶的Co2+的質量/μg,縱坐標表示二階導數波峰電流/nA)。
實施例2 微量(較高濃度)鈷測定體系的線性回歸曲線的制作方法
(1)取Co2+標液(標準溶液)用二次蒸餾水稀釋成適宜濃度的Co2+。
(2)分別量取1.0×10-5、2.0×10-5、4.0×10-5、8.0×10-5、4.0×10-6、1.6×10-4、3.0×10-4、3.2×10-4、4.0×10-4、5.0×10-4和6.0×10-4mol/L Co2+標液各0.10mL于11個50mL容量瓶中,分別向容量瓶中加入2.6mol/L Zn2+0.10mL,丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,靜止5min,分別把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得各濃度Co2+對應的二階導數波峰電流,繪制線性回歸曲線如圖2(橫坐標表示加入容量瓶的Co2+的質量/mg,縱坐標表示二階導數波峰電流/nA)所示。
實施例3 采用體系一測定鋅電解新液中Co2+
(1)量取0.10mL的鋅電解新液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,亞硝酸鈉溶液4.0mL,靜止5min,把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得二階導數波峰電流I1,檢測結果如圖3所示(橫坐標表示掃描電壓/mV,縱坐標表示二階導數波峰電流/nA)。
(2)量取3.0×10-6mol/L Co2+標液0.10mL、0.10mL鋅電解新液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,亞硝酸鈉4.0mL,靜止5min,把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得二階導數波峰電流I2。
(3)鋅電解新液中Co2+的濃度為3.0×10-6I1/(I2–I1)(mol/L)。具體檢測結果數據見表1。
實施例4 采用體系二測定中性上清液中Co2+
(1)量取0.10mL的中性上清液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,靜止5min,把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得二階導數波峰電流I1,檢測結果如圖4所示(橫坐標表示掃描電壓/mV,縱坐標表示二階導數波峰電流/nA)。
(2)量取6.0×10-4mol/L Co2+標液0.10mL、0.10mL中性上清液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,靜止5min,把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得二階導數波峰電流I2。
(3)中性上清液中Co2+的濃度為6.0×10-4I1/(I2–I1)(mol/L)。具體檢測結果數據見表2。
實施例5 采用體系一測定中性上清液中Co2+
(1)量取1.0mL的中性上清液于1000mL容量瓶中,定容,此為待測溶液。配置3.0×10-3mol/L EDTA的氫氧化鈉溶液備用,其余試劑濃度同上。
(2)量取1.0mL步驟(1)待測溶液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入步驟(1)所配EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,亞硝酸鈉4.0mL,靜止5min,把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得二階導數波峰電流I1。檢測結果如圖5所示(橫坐標表示掃描電壓/mV,縱坐標表示二階導數波峰電流/nA)。
(3)量取8.0×10-6mol/L Co2+標液0.10mL,1.0mL的步驟(1)待測溶液于50mL容量瓶中,加入丁二酮肟的乙醇溶液1.4mL,晃動1min,然后依次加入步驟(1)所配EDTA的氫氧化鈉溶液1.0mL,氨-氯化銨緩沖液8.0mL,亞硝酸鈉4.0mL,靜止5min,把溶液倒入電解杯中測定產生的Co2+絡合物吸附極譜波,獲得二階導數波峰電流I2。
(4)中性上清液中Co2+的濃度為8.0×10-6I1/(I2-I1)(mol/L)。具體檢測結果數據見表3。
實驗例1 本發明方法精密度及其與亞硝基R鹽分光光度法(NRS)的比較
1.采用體系一測定鋅電解新液中Co2+濃度的精密度以及與NRS法測定結果的比較
按實施例3方法進行測定,重復5次。本法五次平行測定值如表1所示,相對標準偏差(RSD)1.1%;結果平均值5.80×10-6mol/L。取相同的鋅電解新液樣品按照經典方法-亞硝基R鹽分光光度法(NRS)【標準號:GB/T 223.22-1994】法進行測定,結果見下表1。本發明方法相對于測定鋅電解液中鈷的(NRS)測定結果5.74×10-6mol/L偏高1.05%。
表1采用體系一測定鋅電解新液中Co2+的結果及其精密度,以及與NRS法測定結果的比較(平行測定5次)
2.采用體系二測定中性上清液中Co2+濃度的精密度以及與NRS法測定結果的比較
按實施例4方法進行測定,重復5次。本法五次平行測定值如表2所示,RSD為0.85%;結果平均值6.50×10-4mol/L。取相同的中性上清液樣品按照NRS法進行測定,結果見下表2。本發明方法相對于NRS法測定結果6.57×10-4mol/L偏低1.07%。
表2采用體系二測定中性上清液中Co2+的結果及其精密度,以及與NRS法測定結果的比較(平行測定5次)
3.采用體系一測定中性上清液中Co2+濃度的精密度以及與NRS法測定結果的比較
按實施例5方法進行測定,重復5次。本法五次平行測定值如表3所示,RSD為0.81%;結果平均值6.49×10-4mol/L。取相同的中性上清液樣品按照NRS法進行測定,結果見下表3。本發明方法相對于NRS法測定結果6.51×10-4mol/L偏低0.31%。
表3采用體系一測定中性上清液中Co2+的結果及其精密度,以及與NRS法測定結果的比較(平行測定5次)
由表1、表2和表3中結果可知,該方法兩個體系對測定不同電解鋅液中鈷的相對標準偏差≤1.1%,滿足方法精密度要求;該方法兩個體系測定相同樣品中鈷的相對偏差為0.15%,相對于測定鋅電解液中鈷的經典方法-亞硝基R鹽分光光度法測定結果偏差的絕對值≤1.07%,滿足方法準確度要求。
雖然,上文中已經用一般性說明及具體實施方案對本發明作了詳盡的描述,但在本發明基礎上,可以對之作一些修改或改進,這對本領域技術人員而言是顯而易見的。因此,在不偏離本發明精神的基礎上所做的這些修改或改進,均屬于本發明要求保護的范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!