一種堿性紫1標準物質的制備方法與流程
本發明涉及一種堿性紫1標準物質的制備方法,屬于標準品制備領域。
背景技術:
世界各國對人工合成色素的可用品種、使用量以及使用范圍等都有嚴格的限制。2000年以來,我國共批準使用的合成色素有21種。其中,胭脂紅、莧菜紅、檸檬黃、日落黃、誘惑紅和亮藍等均為常用色素,其使用范圍也有嚴格規定,如禁止用于肉類、魚類、水果及其制品等,僅限用于飲料、配制酒、糖果、糕點和青梅等。其他國家如美國等最早先后批準使用的合成色素達24種,但至今保留僅為9種;日本及歐共體允許使用的有11種。除規定之外的所有人工合成的磺化、硝化、偶氮化染料均不允許以任何形式添加于食品中。然而,在利益的誘惑下,這些具有嚴重危害的添加劑常被不法分子過量添加于食品中,長期食用含有過量合成色素的食品必將嚴重威脅人體健康。所以研究人員開發了多種用于檢測分析食品中化學合成色素含量的技術方法,對這些國家規定限量的添加劑進行定性、定量分析。然而在合成色素非法添加現象無孔不入的今天,快速、準確的檢測方法必然更加能夠為相關部門監管、打擊不法分子提供更為及時有力的依據,同時也為人民群眾的健康提供強有力的技術保障。
堿性紫1,又稱甲基紫,為副品紅堿的四甲基、五甲基、六甲基衍生物的混合物,系綠色發光粉末,熔點137℃。能溶于水和醇,呈紫色溶液,能溶于甘油和氯仿,但不溶于醚。堿性紫1是一種生物染色劑,酸堿指示劑,檢定汞、銀及錫等,密封避光保存。在COMAR數據庫和國家標準物質信息服務平臺數據庫查詢,沒有堿性紫1標準物質的相關信息。鑒于此染料標準物質在食品安全領域的重要作用,發展和研制相關標準物質具有非常重要的現實意義。
技術實現要素:
本發明要解決的技術問題是克服現有技術的缺陷,提供了一種堿性紫1標準物質的制備方法,該方法制備堿性紫1標準物質易操作、簡單快捷,得到的標準品純度高。
為了解決上述技術問題,本發明提供了如下的技術方案:
一種堿性紫1標準物質的制備方法,包括以下各步驟:
(1)供試品溶液的制備:將原料溶于有機溶劑,并采用超聲輔助溶解得堿性紫1原料液,將所堿性紫1原料液經微孔濾膜過濾,調節溶液pH至7.0,然后抽濾獲得供試品溶液;
(2)液相色譜制備:將所述供試品溶液注入制備色譜中分離純餾分,其色譜條件為:
色譜柱為Venusil XBP C18(10μm,50*250mm)、SunFire C18(10μm,50*250mm)或Apollo C18(150×4.6mm,5μm);
流動相為水:乙腈,或甲醇:0.2%TFA水=65:35(v/v);
流動相流速為80mL·min-1;
洗脫方式為梯度洗脫:梯度為0-30min,10%乙腈-90%乙腈(v/v),或運行15min勻速洗脫;
檢測波長254nm;
柱溫30℃;
進樣量0.1-1ml;
接收8-16分鐘內最高色譜峰的餾分,置于預先氮氣填充的外壁包裹錫箔紙的棕色瓶中,然后加入鹽酸中和至pH7;
(3)標準品制備:先在常溫避光狀態下用氮氣吹去全部液態溶劑,然后置于凍干機中凍干,凍干后的堿性紫1固體碾磨粉碎后,再次凍干得堿性紫1標準物質。
在上述方案中優選的是,步驟(1)中所述原料為工業級堿性紫染料,其中堿性紫1的含量在20%-50%。其中可能包含堿性紫1的副品紅堿的四甲基、五甲基、六甲基衍生物及其氧化物、降解物、雜質等。
在上述方案中優選的是,步驟(1)中所述有機溶劑包括純乙腈,體積百分含量為70%—90%的乙腈,體積百分含量為0—20%的甲醇,體積百分含量為0—20%的二氯甲烷,體積百分含量為0—20%的乙酸乙酯。優選為純乙腈。
在上述方案中優選的是,步驟(1)中超聲頻率為20-40Hz。優選為30Hz。
步驟(1)中微孔濾膜優選為0.22μm濾膜。
步驟(1)中優選為用1mol/L的鹽酸調節溶液pH至7.0。
在上述方案中優選的是,步驟(2)中色譜柱為Venusil XBP C18(10μm,50*250mm)。
在上述方案中優選的是,步驟(2)中流動相為水:乙腈。
在上述方案中優選的是,步驟(2)中洗脫方式為梯度洗脫:梯度為0-30min,10%乙腈-90%乙腈(v/v)。
在上述方案中優選的是,步驟(2)中進樣量0.1ml。
在上述方案中優選的是,步驟(3)中標準品制備:先在常溫避光狀態下用氮氣吹去全部液態溶劑,然后置于凍干機-50℃中凍干12-24小時,凍干后的堿性紫1固體碾磨粉碎后,再次凍干12-24小時。
本發明所述的堿性紫1標準物質用于食品中非法色素添加的定性定量檢測。
現在工業上主要是通過改進合成方法提高由堿性品紅合成為堿性紫的收率來提高產品純度。目前商品化堿性紫1純度約20%-80%。
本發明的有益效果:本發明的方法制備堿性紫1標準物質純度高,可用于食品中非法色素添加的定性定量檢測。本發明的方法工藝簡單、操作方便、分離速度快,制備的堿性紫1純度高、產品純度可達到99.3%,產品質量穩定,均勻性好,流動相可回收利用,無污染,實現清潔生產。
附圖說明
圖1為堿性紫1的紅外光譜圖;
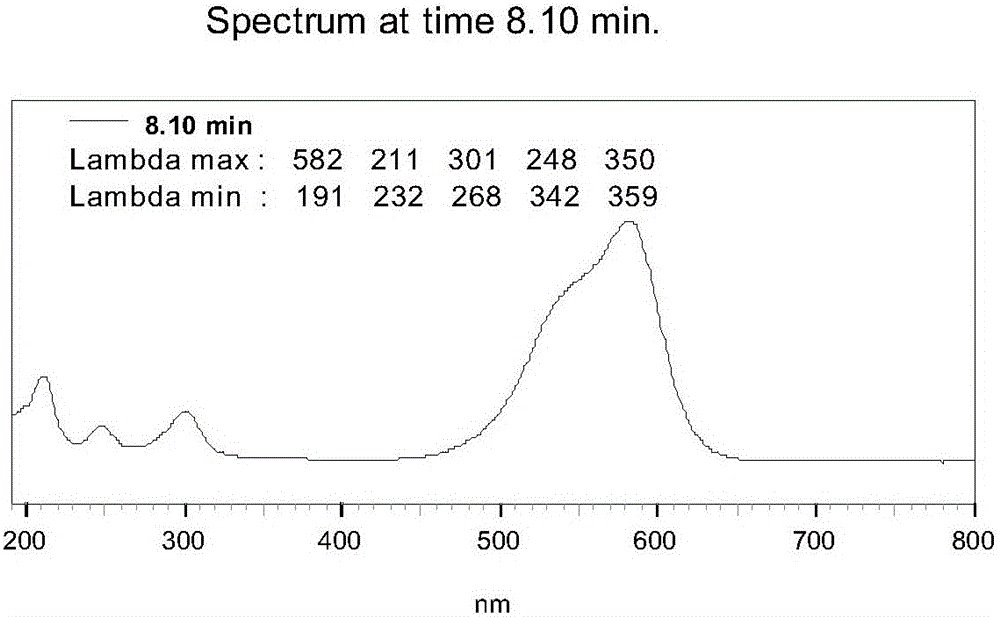
圖2為堿性紫1的紫外可見光吸收光譜圖;
圖3為堿性紫1的一級質譜圖;
圖4為堿性紫1的二級質譜圖;
圖5為堿性紫1的1H-NMR譜圖;
圖6為堿性紫1的13C-NMR譜圖。
具體實施方式
以下對本發明的優選實施例進行說明,應當理解,此處所描述的優選實施例僅用于說明和解釋本發明,并不用于限定本發明。
實施例1:
一種堿性紫1標準物質的制備方法,包括以下各步驟:
(1)供試品溶液的制備:將含量約為20%的堿性紫1原料溶于甲醇并采用30Hz超聲溶解,經0.22μm濾膜過濾,調節溶液pH至7.0,然后抽濾獲得供試品溶液;;
(2)液相色譜制備:將堿性紫1供試品溶液注入制備色譜分離純餾分,其色譜條件為:
色譜柱為SunFire C18(10μm,50*250mm),
流動相為水:乙腈,
流動相流速為80mL·min-1,
洗脫方式為梯度洗脫:梯度為0-30min,10%乙腈-90%乙腈(v/v),
檢測波長254nm,
柱溫30℃,
進樣量0.5ml,
收集14.9min餾分接收于預先氮氣填充的外壁包裹錫箔紙的棕色瓶中,然后加入少量鹽酸中和至pH7;
(3)標準品制備:先在常溫避光狀態下用旋轉蒸發儀去全部液態溶劑,獲得的堿性紫1固體純度為97.4%。
本實施例得到的堿性紫1的紅外光譜圖如圖1,紫外可見光吸收光譜見圖2,一級質譜見圖3,二級質譜見圖4,1H-NMR譜見圖5(399.65MHz;0.032g:0.5ml DMSO-d6),13C-NMR(100.54MHz;0.032g:0.5ml DMSO-d6)譜見圖6。
實施例2:
一種堿性紫1標準物質的制備方法,包括以下各步驟:
(1)供試品溶液的制備:將含量約為20%的堿性紫1原料溶于純乙腈并采用30Hz超聲溶解,經0.22μm濾膜過濾,用1mol/L的鹽酸調節溶液pH至7.0,然后抽濾獲得供試品溶液;;
(2)液相色譜制備:將堿性紫1供試品溶液注入制備色譜分離純餾分,其色譜條件為:
色譜柱為Apollo C18(150×4.6mm,5μm);
流動相為甲醇:0.2%TFA水=65:35(v/v);
流速為1.0mL·min-1;
柱溫為30℃;
運行時間為15min;
檢測波長為254nm;
柱溫為30℃;
進樣量為0.1ml,
收集8.7min餾分接收于預先氮氣填充的外壁包裹錫箔紙的棕色瓶中,然后加入少量鹽酸中和至pH7;
(3)標準品制備:先在常溫避光狀態下用氮氣吹去全部液態溶劑,然后置于凍干機-50℃中凍干24小時,凍干后的堿性紫1固體純度為96.7%。
實施例3:
一種堿性紫1標準物質的制備方法,包括以下各步驟:
(1)供試品溶液的制備:將含量約為20%的堿性紫1原料溶于純乙腈,并采用30Hz超聲溶解,經0.22μm濾膜過濾,用1mol/L的鹽酸調節溶液pH至7.0,然后抽濾獲得供試品溶液;
(2)液相色譜制備:將堿性紫1供試品溶液注入制備色譜分離純餾分,其色譜條件為:
色譜柱為Venusil XBP C18(10μm,50*250mm),
流動相為水:乙腈,
流動相流速為80mL·min-1,
洗脫方式為梯度洗脫:梯度為0-30min,10%乙腈-90%乙腈(v/v),
檢測波長254nm,
柱溫30℃,
進樣量0.1ml,
收集13.6min餾分接收于預先氮氣填充的外壁包裹錫箔紙的棕色瓶中,然后加入少量鹽酸中和至pH7;
(3)標準品制備:先在常溫避光狀態下用氮氣吹去全部液態溶劑,然后置于凍干機-50℃中凍干24小時,凍干后的堿性紫1固體純度為99.3%。
實施例4:
一種堿性紫1標準物質的制備方法,包括以下各步驟:
(1)供試品溶液的制備:將含量約為20%的堿性紫1原料溶于純乙腈,并采用30Hz超聲溶解,經0.22μm濾膜過濾,調節溶液pH至7.0,然后抽濾獲得供試品溶液;;
(2)液相色譜制備:將堿性紫1供試品溶液注入制備色譜分離純餾分,其色譜條件為:
色譜柱為SunFire C18(10μm,50*250mm),
流動相為水:乙腈,
流動相流速為80mL·min-1,
洗脫方式為梯度洗脫:梯度為0-30min,10%乙腈-90%乙腈(v/v),
檢測波長254nm,
柱溫30℃,
進樣量0.5ml,
收集14.9min餾分接收于預先氮氣填充的外壁包裹錫箔紙的棕色瓶中,然后加入少量鹽酸中和至pH7;
(3)標準品制備:先在常溫避光狀態下用旋轉蒸發儀去全部液態溶劑,獲得的堿性紫1固體純度為98.1%。
實施例5:
一種堿性紫1標準物質的制備方法,包括以下各步驟:
(1)供試品溶液的制備:將含量約為20%的堿性紫1原料溶于純乙腈并采用30Hz超聲溶解,經0.22μm濾膜過濾,用1mol/L的鹽酸調節溶液pH至7.0,然后抽濾獲得供試品溶液;
(2)液相色譜制備:將堿性紫1供試品溶液注入制備色譜分離純餾分,其色譜條件為:
色譜柱為Apollo C18(150×4.6mm,5μm);
流動相為水:乙腈;
流動相流速為80mL·min-1;
洗脫方式為梯度洗脫:梯度為0-30min,10%乙腈-90%乙腈(v/v);
流速為80mL·min-1;
柱溫為30℃;
檢測波長為254nm;
柱溫30℃;
進樣量0.1ml;
收集8.7min餾分接收于預先氮氣填充的外壁包裹錫箔紙的棕色瓶中,然后加入少量鹽酸中和至pH7;
(3)標準品制備:先在常溫避光狀態下用氮氣吹去全部液態溶劑,然后置于凍干機-50℃中凍干24小時,凍干后的堿性紫1固體純度為97.2%。
最后應說明的是:以上所述僅為本發明的優選實施例而已,并不用于限制本發明,盡管參照前述實施例對本發明進行了詳細的說明,對于本領域的技術人員來說,其依然可以對前述各實施例所記載的技術方案進行修改,或者對其中部分技術特征進行等同替換。凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!