六方氮化硼納米片在制備基質溶液中的用途以及質譜檢測方法與流程
本發明涉及質譜分析領域,具體地,本發明涉及采用基質輔助激光解吸/電離質譜分析小分子的方法。
背景技術:
分子量是有機化合物最基本的理化性質參數。分子量正確與否往往代表著所測定的有機化合物及生物大分子的結構正確與否。基質輔助激光解吸/電離質譜(MALDI-MS)是近年來的一種新型軟電離生物質譜,基質輔助激光解吸/電離(MALDI)的原理是用激光照射基質與樣品分子形成的共結晶薄膜,基質從激光中吸收能量傳遞給樣品分子,將質子傳遞給樣品分子或從樣品分子中得到質子,從而實現樣品分子的電離,可以不產生或產生較少的碎片離子。通常基質輔助激光解析/電離技術采用飛行時間質譜(TOF-MS)作為質量分析手段。它可直接應用于混合物的分析,也可用來檢測樣品中是否含有雜質及雜質的分子量。分子量也是生物大分子如多肽、蛋白質等鑒定中首要的參數,也是基因工程產品報批的重要數據之一。MALDI-TOF的準確度高達0.1%~0.01%,遠遠高于目前常規應用的聚丙烯酰胺凝膠(SDS)電泳與高效凝膠色譜技術。MALDI-TOF-MS具有操作簡單、快速、譜圖直觀、能耐受一定濃度的鹽和去垢劑等特點,特別適合于混合多肽、蛋白、寡核苷酸的精確質量數測定,其測定質量數范圍最高可達40萬Da以上,靈敏度可達10-15~10-18摩爾,質量準確度可達5ppm。MALDI-TOF-MS儀器主要由兩部分組成:基質輔助激光解吸/電離離子源(MALDI)和飛行時間質量分析器(TOF)。TOF的原理是離子在電場作用下加速飛過飛行管道,根據到達檢測器的飛行時間不同而被檢測即測定離子的質荷比(m/z)與離子的飛行時間成正比,從而完成對待測物質的檢測。MALDI-TOF-MS具有靈敏度高、準確度高及分辨率高等特點,為生命科學等領域提供了一種強有力的分析測試手段,并正扮演著越來越重要的作用。然而由于基質輔助激光解吸/電離飛行時間質譜(MALDI-TOF MS)常用的基質(例如α-腈基-4-羥基肉桂酸(CHCA)、2,5-二羥基苯甲酸(DHB)、芥子酸(SA)、3-羥基-2-吡啶甲酸(3-HPA)、蒽三酚(DI)以及3-氨基喹啉(3-AQ)等)在分析過程中發生碎裂及分子之間的締合等,常常會在質荷比(m/z)小于1000的范圍內產生嚴重的基質背景干擾現象,因此嚴重影響分析質荷比小于1000的小分子化合物的效果。目前,納米材料(包括納米金、納米銀、碳量子點、石墨烯、TiO2、TiN、Si)在激光解吸/電離離子源 質譜分析中已經可作為基質應用,輔助分析多種有機化合物。
然而,用于基質輔助激光解吸/電離質譜的基質以及基于基質輔助激光解吸/電離質譜分析小分子的方法仍有待改進。
技術實現要素:
本申請是基于發明人對以下事實和問題的發現和認識作出的:
盡管納米材料基質相對于傳統有機基質,已經極大程度地減小了低分子量背景干擾,但復雜的制備和昂貴的成本令這一方法難以普及。因此,尋找一種操作制備簡單、成本低廉、靈敏度高、在檢測范圍內沒有背景干擾的基質,對當前MALDI-MS分析具有重要的意義。發明人經過深入研究以及大量實驗發現,六方氮化硼(h-BN)納米片在兼具納米基質材料優點的同時,具有較高的熱傳導性以及穩定性,高抗氧化性以及良好的化學穩定性。六方氮化硼納米片,即所謂的“白石墨烯”,由數層六方氮化硼構成。它具有獨特的物化性質,例如高的能帶寬、良好的電絕緣性、具有紫外光致發光特性、高熱傳導性以及良好的穩定性(真空中超過3000開)等。并且,h-BN納米片具有的高比表面積以及B-N鍵的極性有利于h-BN納米片與其他分子發生如相互作用,并且能夠提供良好的吸附性能。此外,發明人經過大量實驗證實,h-BN納米片在低質量范圍內,即質荷比(m/z)為0~1500范圍內完全沒有背景干擾。因此,可以將h-BN納米片作為基質,用于MALDI-MS。
本發明旨在至少在一定程度上解決相關技術中的技術問題之一。為此,本發明提出一種采用六方氮化硼納米片作為基質的MALDI-MS分析小分子的方法。采用該方法分析質荷比為0~1500范圍內的小分子,分析結果不受基質的背景峰干擾,因此提高了分析結果的準確性。
在本發明的一個方面,本發明提出了六方氮化硼納米片在制備基質溶液中的用途,所述基質溶液用于基質輔助激光解吸/電離質譜檢測。由此,可以避免基質在分析過程中產生背景峰進而對樣品的檢測造成干擾,從而提高了質譜檢測的準確性。
在本發明的另一方面,本發明提出了一種樣品檢測的方法。根據本發明的實施例,該方法包括:(1)將基質溶液與樣品混合,所述基質溶液含有六方氮化硼納米片;以及(2)將步驟(1)中所得到的混合物加入到基質輔助激光解吸/電離質譜儀中,以便對所述樣品進行基質輔助激光解吸/電離質譜檢測。由此,可以采用含有六方氮化硼納米片的基質溶液輔助待測物進行電離,進而可以提高質譜檢測的效果以及檢測結果的準確性。
根據本發明的實施例,所述樣品為質荷比為0~1500的分子。由此,可以利用根據本發明實施例的方法,實現上述樣品的質譜檢測,進而擴展該方法的應用范圍。
根據本發明的實施例,所述樣品是生物組織切片,并且所述方法進一步包括:(3)基 于所述基質輔助激光解吸/電離質譜檢測的結果,構建所述生物組織切片的圖像。由此,可以利用該方法實現質譜成像分析,進而可以擴展該方法的應用范圍。
根據本發明的實施例,在該方法中,步驟(1)進一步包括:(1-1)將所述六方氮化硼納米片與溶劑混合,以便獲得所述基質溶液,其中,所述六方氮化硼納米片的濃度為0~1mg/ml,所述溶劑包括選自水、甲醇、乙醇以及乙腈的至少之一,任選地,所述六方氮化硼納米片的濃度為0.1~1mg/ml;以及(1-2)將所述樣品與所述基質溶液等體積混合,其中,所述樣品與所述基質溶液的濃度比為(1~5mmol/L):(0~1mg/ml),任選地,所述樣品與所述基質溶液的濃度比為1mmol/L:0.25mg/ml。其中,當基質溶液與樣品溶液的混合物中基質溶液的含量過低時,無法有效地輔助樣品發生電離,而當基質溶液含量過高時,則會造成基質的浪費。由此,當樣品與基質溶液的濃度比例為上述范圍內時,可以保證樣品的有效電離同時不會造成基質溶液的浪費,進而進一步提高質譜檢測的檢測效果。
在本發明的又一方面,本發明提出了一種通過質譜法檢測質荷比為0~1500的小分子的方法。根據本發明的實施例,該方法包括:(1)將六方氮化硼納米片與溶劑混合,以便獲得基質溶液,其中,所述六方氮化硼納米片的濃度為0~1mg/ml,所述溶劑包括選自水、甲醇、乙醇以及乙腈的至少之一,并且,將所述小分子與所述溶劑混合,以便獲得小分子溶液,所述小分子溶液的濃度為1mmol/L;將所述基質溶液與所述小分子溶液按照體積比為1:1進行混合,以便形成混合樣品;(2)將1μl所述混合樣品加入到靶板上,并在空氣中干燥,以便除去所述溶劑;以及(3)將步驟(2)中獲得的所述靶板放入質譜儀中,以便完成所述質譜檢測,其中,所述質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,所述質譜檢測采用負離子模式,所述質譜檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次。由此,可以利用該方法完成質荷比為0~1500的小分子的質譜檢測,并避免基質在分析過程產生背景峰而對分析結果造成影響,進而提高了該方法的檢測效果。
在本發明的又一方面,本發明提出了一種通過質譜法檢測水體污染物的方法。根據本發明的實施例,該方法包括:(1)將六方氮化硼納米片與溶劑混合,以便獲得基質溶液,其中,基于所述基質溶液的總體積,所述六方氮化硼納米片的濃度為0~1mg/ml,所述溶劑包括選自水、甲醇、乙醇以及乙腈的至少之一,并且,將污水與體積比為1:1的甲醇與水的混合物進行混合,配置水體污染物待測液,將所述基質溶液與所述水體污染物待測液混合,以便形成混合樣品;(2)將1μl所述混合樣品加入到靶板上,并在空氣中干燥;以及(3)將步驟(2)中獲得的所述靶板放入質譜儀中,以便完成所述質譜檢測,其中,所述質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,所述質譜檢測采用負離子模式,所述質 譜檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次。由此,可以利用該方法完成復雜體系中水體污染物的質譜檢測,并避免基質在分析過程產生背景峰而對分析結果造成影響,進而提高了該方法的檢測效果。
在本發明的又一方面,本發明提出了一種通過質譜法檢測微生物代謝產物的方法。根據本發明的實施例,該方法包括:(1)將所述微生物培養到OD值為0.8~1.0時收集,采用磷酸鹽緩沖溶液洗滌所述微生物,將經過所述洗滌的所述微生物在9000g條件下離心,以便獲得沉淀,取10mg所述沉淀,并向10mg所述沉淀中加入體積分數為80%的甲醇水溶液進行混合,加入的所述甲醇水溶液的體積為10mg所述沉淀的3倍,靜置,以便獲得微生物樣品;(2)將六方氮化硼納米片與溶劑混合,以便獲得基質溶液,其中,基于所述基質溶液的總體積,所述六方氮化硼納米片的濃度為0~1mg/ml,所述溶劑包括選自水、甲醇、乙醇以及乙腈的至少之一,并將所述微生物樣品與所述基質溶液混合,以便獲得待測樣品;(3)將所述待測樣品滴加到靶板上,并在空氣中干燥;以及(4)將步驟(3)獲得的所述靶板放入質譜儀中,以便完成所述質譜法檢測,其中,所述質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,所述質譜法檢測采用負離子模式,所述質譜法檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次。由此,可以利用該方法在非均質體系中完成微生物代謝產物的質譜檢測,并避免基質在分析過程產生背景峰而對分析結果造成影響,進而提高了該方法的檢測效果。
在本發明的又一方面,本發明提出了一種對組織切片進行質譜成像分析的方法。根據本發明的實施例,該方法包括:(1)將冷凍的組織在冷凍切片機上進行切片,以便獲得所述組織切片,其中,所述組織切片的厚度為5μm-50μm;(2)將六方氮化硼納米片與溶劑混合,以便獲得基質溶液,其中,基于所述基質溶液的總體積,所述六方氮化硼納米片的濃度為0~1mg/ml,所述溶劑包括選自水、甲醇、乙醇以及乙腈的至少之一;(3)將所述組織切片與所述基質溶液進行混合并貼至ITO(氧化銦錫)玻璃表面,并干燥,以便獲得待測樣品;以及(4)將所述待測樣品放入質譜儀中,以便完成所述質譜成像分析,其中,所述質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,所述質譜成像分析采用負離子模式,所述質譜法檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次,掃描分辨率為100μm,激光光斑為50μm,成像數據采用FlexImaging v3.0或者BioMap v3.8處理。由此,可以利用該方法完成組織切片的質譜成像分析,并避免基質在分析過程產生背景峰而對分析結果造成影響,進而提高了該方法的檢測效果。
附圖說明
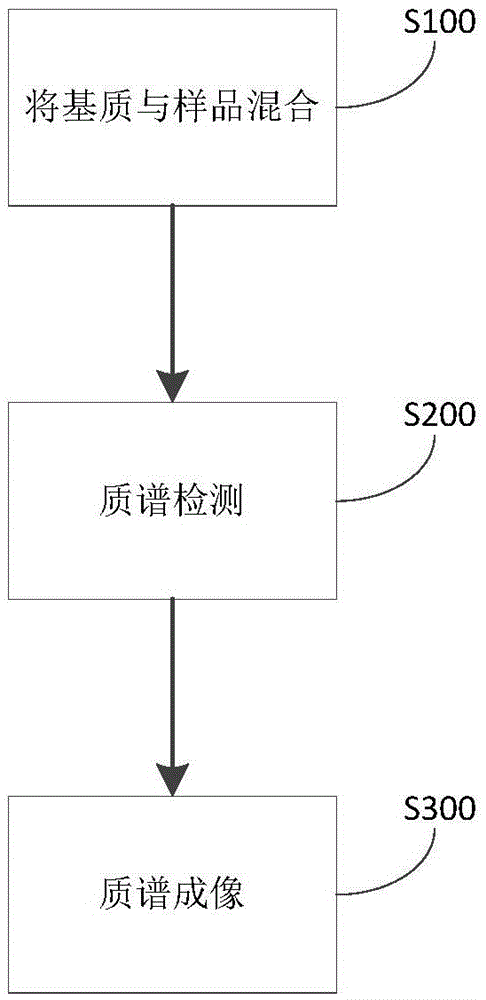
圖1顯示了根據本發明一個實施例的樣品檢測方法的流程圖;
圖2顯示了根據本發明另一個實施例的樣品檢測方法的流程圖;
圖3顯示了根據本發明實施例1的含有六方氮化硼納米片的基質溶液的基質輔助激光解吸/電離飛行時間質譜檢測圖;
圖4顯示了根據本發明實施例2的氨基酸基質輔助激光解吸/電離飛行時間質譜檢測圖;
圖5顯示了根據本發明實施例3的絲氨酸、苯丙氨酸富集前后的基質輔助激光解吸/電離飛行時間質譜檢測圖;
圖6顯示了根據本發明實施例4的水體污染物9-硝基蒽富集前后的基質輔助激光解吸/電離飛行時間質譜檢測圖;
圖7以及圖8顯示了根據本發明實施例5的大腸桿菌代謝產物的基質輔助激光解吸/電離飛行時間質譜檢測圖;以及
圖9以及圖10顯示了根據本發明實施例6的小鼠腦組織質譜成像檢測圖。
具體實施方式
下面詳細描述本發明的實施例,所述實施例的示例在附圖中示出。下面通過參考附圖描述的實施例是示例性的,旨在用于解釋本發明,而不能理解為對本發明的限制。
在本發明的一個方面,本發明提出了六方氮化硼納米片在制備基質溶液中的用途,上述基質溶液用于基質輔助激光解吸/電離質譜檢測。根據本發明的實施例,將六方氮化硼納米片加入到水、甲醇、乙醇以及乙腈或者上述物質形成的互溶體系中,配置含有六方氮化硼納米片的基質溶液,并用于基質輔助激光解吸/電離質譜的檢測。由此,可以避免基質在檢測過程中在質荷比為0~1500范圍內出現背景峰,進而提高基質輔助激光解吸/電離質譜檢測的效果。
在本發明的另一方面,本發明提出了一種樣品檢測的方法。根據本發明的實施例,參考圖1,該方法包括:
S100:將基質與樣品混合
根據本發明的實施例,在該步驟中,將含有六方氮化硼納米片的基質溶液與待測樣品混合。需要說明的是,在本發明中,配置基質溶液的溶劑以及六方氮化硼納米片相對于基質溶液總體積的具體濃度不受特別限制,本領域技術人員可以根據實際情況,例如,根據待測樣品的濃度以及種類,選擇適當的物質作為溶劑,配置具有一定濃度的含有六方氮化硼納米片的基質溶液。本領域技術人員能夠理解,只要選擇的溶劑能夠與后續的質譜分析 兼容,并且基質溶液中六方氮化硼納米片的含量能夠滿足吸收激光并將能量傳遞給待測物質,使足夠的待測物被離子化即可。具體地,根據本發明的實施例,基質溶液可以按照以下方法制備:選擇水、甲醇、乙醇以及乙腈的至少之一,或者含有上述物質組成的互溶體系作為溶劑,將六方氮化硼納米片納米片與溶劑混合,配置基質溶液。需要說明的是,在本發明中,術語“互溶體系”特指含有水、甲醇、乙醇以及乙腈的至少之一的、體系中各組分可按照任意比例互溶的液體混合物。并且,在基質溶液中,六方氮化硼納米片的濃度可以為0~1mg/ml(不包含0)。例如,根據本發明的一些實施例,六方氮化硼納米片的濃度可以為0.1~1mg/ml。由此,可以獲得具有適當濃度并且與后續質譜檢測相兼容的含有六方氮化硼納米片的基質溶液,進而可以提高后續質譜檢測的檢測效果,并且避免由于基質溶液在質譜檢測的過程中產生背景峰而對檢測結果造成的負面影響。
根據本發明的實施例,在該步驟中,將待測樣品與基質溶液等體積進行混合,待測樣品與基質溶液的濃度比可以為(1~5mmol/L):(0~1mg/ml)。由此,可以待測樣品在基質溶液的輔助下更好地發生電離。優選地,待測樣品與基質溶液的濃度比可以為1mmol/L:0.25mg/ml。由此,可以在保證待測樣品能夠高效地發生電離的同時,避免添加過多的基質溶液而造成浪費,進而可以進一步提高該方法的檢測效率以及檢測效果。
根據本發明的實施例,在本發明中,待測樣品可以為質荷比為0~1500的分子。需要說明的是,待測樣品的種類并不受特別限制,只要能夠適用于采用MALDI-MS方法進行質譜檢測的物質均可作為本發明的待測樣品。本領域技術人員可以選擇適用于采用MALDI技術進行電離的物質作為待測樣品,以便更好地發揮根據本發明實施例的方法的特點以及優勢。例如,根據本發明的一些實施例,待測樣品可以為氨基酸、維生素、有機酸、多肽、甾體、有機污染物、硫苷脂或者為上述物質構成的混合物。在本發明中個,術語“有機污染物”包含但不限于常見的水體中的污染物。例如,根據本發明的一些實施例,有機污染物可以包含氨基酸、蛋白質、油類以及脂類等耗氧有機物、多環芳烴類物質、有機磷農藥、有機氯農藥等物質。由此,可以更好地發揮該方法在質譜檢測中的特征以及優點,進而可以高效準確地對待測樣品進行分析。
S200:質譜檢測
根據本發明的實施例,在該步驟中,將含有待測樣品以及基質溶液的混合物加入到基質輔助激光解吸/電離質譜儀中,以便對樣品進行質譜檢測。具體地,根據本發明的實施例,可以將含有待測樣品以及基質溶液的混合物滴加到靶板或者玻璃板上,在空氣中進行干燥以便通過揮發除去混合物中的溶劑。然后,將含有待測樣品以及六方氮化硼納米片基質的靶板或者玻璃板送入質譜儀中,完成對待測樣品的質譜檢測。
根據本發明的實施例,在利用該方法對樣品進行檢測,當樣品為生物組織切片時,參 考圖2,該方法進一步包括:
S300:質譜成像
根據本發明的實施例,在該步驟中,基于基質輔助激光解吸/電離質譜檢測的結果,構建生物組織切片的圖像。具體地,在該步驟中,對生物組織切片做逐點掃描的質譜檢測,以便獲得生物組織切片中每一像素點的化學成分,并基于同一化學成分,構建該成分在生物組織切片中的分布圖。由此,可以利用該方法實現對組織切片中特定成分分布的檢測,進而可以進一步提高該方法的應用范圍。
此外,需要說明的是,本發明以六方氮化硼納米片作為MALDI的基質的質譜分析法,通常是用飛行時間質譜(TOF-MS)作為質量分析手段,但與其他質譜質量分析器也兼容。
根據本發明的實施例,由于六方氮化硼納米片在檢測m/z值在0~1500范圍內的分子時,完全不產生背景干擾,因此可以提高采用該方法對上述分子進行質譜檢測時的檢測效果。此外,采用六方氮化硼納米片作為基質可以在正離子和負離子兩種模式下對不同類型的小分子進行檢測,由于氮化硼自身的理化性質,該方法在負離子模式下能夠獲得更加優異的檢測效果。并且,由于六方氮化硼納米片本身容易獲得質子,因此在質譜檢測過程中無需加入離子化試劑,進而降低了利用該方法進行質譜檢測時對于樣品處理的要求。
此外,根據本發明的實施例,采用本發明的方法對痕量物質進行富集檢測實驗時,氮化硼由于其具有高表面積富電子疏水性質,可作為固相微萃取的材料,實現富集檢測與輔助解吸電離的雙重作用,因此可以用于對樣品中的痕量成分進行分析。
在本發明的又一方面,本發明提出了一種通過質譜法檢測質荷比為0~1500的小分子的方法。根據本發明的實施例,上述質荷比為0~1500的小分子可以為質荷比在0~1500的氨基酸、短肽(肽鍵數不超過10個)、維生素、有機酸以及甾體。具體地,根據本發明的實施例,該方法包括:
(1)將小分子與溶劑混合,配置小分子溶液。具體地,根據本發明的實施例,選擇水、甲醇、乙醇以及乙腈的至少之一,或者包括上述物質組成的混溶體系作為溶劑,將一定量的小分子加入到上述溶劑中,配置小分子濃度為1mmol/L的小分子溶液;并且,將一定量的六方氮化硼納米片加入到前面描述的溶劑中,配置基質溶液。根據本發明的實施例,基質溶液的濃度可以為0~1mg/ml(不包含0mg/ml),例如,根據本發明的實施例,基于基質溶液的總體積,六方氮化硼納米片的濃度為0.1mg/ml。然后,將前面配置的小分子溶液與基質溶液按照體積比為1:1進行混合,以便獲得混合樣品。由此,可以獲得具有適當小分子含量以及六方氮化硼納米片含量的混合樣品,進而可以更好地使小分子電離,從而提高后續質譜檢測的檢測效果。
(2)根據本發明的實施例,在該步驟中,將1μl混合樣品加入到靶板上,并在空氣中 干燥,以便除去混合樣品中的溶劑。由此,可以在該步驟中除去混合樣品中的溶劑,進而提高后續質譜檢測的檢測效果。
(3)將含有混合樣品的所述靶板放入質譜儀中,以便完成所述質譜檢測。具體地,根據本發明的實施例,質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,采用負離子模式,質譜檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次。由此,可以在不產生背景峰的前提下,完成對小分子的質譜檢測,進而可以調高質譜檢測的檢測效果。
需要說明的是,由于在該方法中采用了含有六方氮化硼納米片的基質溶液,因此前面描述的樣品檢測方法中的特征以及優點同樣適用于通過質譜法檢測質荷比為0~1500的小分子的方法,在此不再贅述。
在本發明的另一方面,本發明提出了一種通過質譜法檢測水體污染物的方法。需要說明的是,在本發明中個,術語“水體污染物”包含但不限于常見的水體中的污染物。例如,根據本發明的一些實施例,水體污染物可以包含硫化物、氟化物、氨基酸、蛋白質、油類以及脂類等耗氧有機物、多環芳烴類物質、有機磷農藥、有機氯農藥等物質。具體地,根據本發明的實施例,該方法包括:
(1)選擇選自水、甲醇、乙醇以及乙腈的至少之一,或者由上述物質組成的混溶體系作為溶劑,將六方氮化硼納米片與溶劑混合,以便獲得基質溶液,其中,基于基質溶液的總體積,六方氮化硼納米片的濃度可以為0~1mg/ml(不包含0mg/ml);并且,將污水與體積比為1:1的甲醇與水的混合物進行混合,配置水體污染物待測液,將基質溶液與水體污染物待測液混合,以便形成混合樣品。需要說明的是,配置的水體污染物待測液可以取上清液部分與基質溶液進行混合,也可以不對水體污染物待測液進行處理,直接取渾濁的水體污染物待測液與基質溶液進行混合。由此,可以獲得具有適當污水以及六方氮化硼納米片含量的混合樣品,進而可以更好地使水體污染物中的待測物電離,從而提高后續質譜檢測的檢測效果。
(2)將1μl混合樣品加入到靶板上,并在空氣中干燥。由此,可以在該步驟中除去混合樣品中的溶劑,進而提高后續質譜檢測的檢測效果。
(3)將含有混合樣品的靶板放入質譜儀中,以便完成所述質譜檢測。其中,根據本發明的實施例,質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,采用負離子模式,質譜檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次。由此,可以在不產生背景峰的前提下,在復雜體系中完成對水體中污染物的質譜檢測,進而可以調 高質譜檢測的檢測效果。
需要說明的是,由于在該方法中采用了含有六方氮化硼納米片的基質溶液,因此前面描述的樣品檢測方法中的特征以及優點同樣適用于通過質譜法檢測水體污染物的方法,在此不再贅述。
在本發明的又一方面,本發明提出了一種通過質譜法檢測微生物代謝產物的方法。根據本發明的實施例,該方法包括:
(1)將微生物培養到OD值為0.8~1.0時收集,采用磷酸鹽緩沖溶液洗滌微生物,將經過洗滌的所述微生物在9000g條件下離心,以便獲得沉淀,取10mg沉淀,并向其中加入體積分數為80%的甲醇水溶液進行混合,加入的所述甲醇水溶液的體積為10mg沉淀體積的3倍。然后靜置,以便獲得微生物樣品。
(2)選擇選自水、甲醇、乙醇以及乙腈的至少之一,或者由上述物質組成的混溶體系作為溶劑,將六方氮化硼納米片與溶劑混合,配置基質溶液。其中,基于基質溶液的總體積,六方氮化硼納米片的濃度為0~1mg/ml(不包括0)。并將步驟(1)中獲得的微生物樣品與基質溶液混合,以便獲得待測樣品。
(3)將待測樣品滴加到靶板上,并在空氣中干燥。
(4)將含有待測樣品的靶板放入質譜儀中,以便完成所述質譜法檢測。其中,根據本發明的實施例,質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,采用負離子模式,質譜法檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次。由此,可以在簡便有效地檢測微生物代謝產物,并且,利用根據本發明實施例的方法進行的質譜檢測不會在檢測過程中出現背景峰,進而可以進一步提高微生物代謝產物檢測的準確性。
在本發明的又一方面,本發明提出了一種對組織切片進行質譜成像分析的方法。根據本發明的實施例,該方法包括:
(1)將冷凍的組織在冷凍切片機上進行切片,以便獲得組織切片。根據本發明的實施例,該組織切片的厚度可以為5μm-50μm。
(2)選擇選自水、甲醇、乙醇以及乙腈的至少之一,或者由上述物質組成的混溶體系作為溶劑,將六方氮化硼納米片與溶劑混合,配置基質溶液。其中,基于基質溶液的總體積,六方氮化硼納米片的濃度為0~1mg/ml(不包括0)。并將步驟(1)中獲得的微生物樣品與基質溶液混合,以便獲得待測樣品。
(3)將組織切片與基質溶液進行混合并貼至ITO(氧化銦錫)玻璃表面,(即具有氧化銦錫層的玻璃表面,其中,組織切片貼在氧化銦錫層的表面)并干燥,以便獲得待測樣品。根據本發明的實施例,在該步驟中,組織切片與基質溶液進行混合的具體方式不受特 別限制。例如,根據本發明的一些實施例,可以先將基質溶液噴灑在組織切片表面,然后將含有基質溶液的組織切片貼合在ITO玻璃表面;也可以現將基質溶液噴灑到ITO玻璃表面,再將組織切片貼合至噴灑了基質溶液的ITO玻璃上;此外,還可以預先在ITO玻璃表面噴灑基質溶液后將組織切片貼合至ITO玻璃表面,然后再在組織切片的上表面再噴灑一層基質溶液。
(4)將步驟(3)中獲得的含有組織切片的ITO玻璃放入質譜儀中,以便完成所述質譜成像分析。根據本發明的實施例,質譜儀為基質輔助激光解吸/電離飛行時間質譜儀,采用負離子模式,質譜法檢測條件為:加速電壓為19.000kv,延遲引出電壓為14.920kv,反射器電壓為20.000kv,透鏡電壓為7.000kv,頻率為1.000Hz,激光器能量為75-80%,累加次數為90次,掃描分辨率為100μm,激光光斑為50μm,成像數據采用FlexImaging v3.0或者BioMap v3.8處理。由此,可以在簡便有效地實現對于組織切片的質譜成像檢測,并且,利用根據本發明實施例的方法進行的質譜檢測不會在檢測過程中出現背景峰,進而可以進一步提高對于組織切片進行質譜成像分析的準確性。
下面通過具體的實施例對本發明進行說明,本領域技術人員能夠理解的是,下面的具體的實施例僅僅是為了說明的目的,而不以任何方式限制本發明的范圍。另外,在下面的實施例中,除非特別說明,所用的基質輔助激光解吸/電離飛行時間質譜儀的型號為BIFLEXTM III(Bruker)和UltrafleXtreme(Bruker)。所采用的材料和設備均是市售可得的。如果在后面的實施例中,未對具體的處理條件和處理方法進行明確描述,則可以采用本領域中公知的條件和方法進行處理。
實施例1基質溶液背景峰測試
采用水作為溶劑,配置六方氮化硼納米片濃度為0.25mg/ml的基質溶液。質譜條件為:電壓:加速電壓:19.000kv;延遲引出電壓:14.920kv;反射器電壓:20.000kv;透鏡電壓:7.000kv;頻率:1.000Hz;激光器能量:75-80%;累加次數:90次;負離子模式。參考圖3,在m/z值0~1000范圍內,沒有基質相關的質譜信號。因此,采用含有六方氮化硼納米片的基質溶液對測試樣本,包括復雜體系進行質譜檢測,不會造成干擾。
實施例2小分子質譜分析
采用含有六方氮化硼納米片的基質溶液對含有不同小分子的溶液進行質譜分析。
分別取配制好的濃度均為1mmol/L的色氨酸溶液(Trp)(圖4A)亮氨酸溶液(Leu)、(圖4B)、天冬氨酸溶液(Asn)(圖4C)、谷氨酸溶液(Glu)(圖4D)、精氨酸溶液(Arg)(圖4E)各1μL,分別與含有六方氮化硼納米片(濃度為0.1mg/mL)的基質溶液1μl進行混合。上述溶液的溶劑均為體積比為1:1的甲醇水溶液。將上述含有氨基酸的溶液分別與基質溶液混合均勻,然后分別向MALDI靶板加入1μL混合樣品,空氣中干燥后進入質譜 分析。質譜條件為:電壓:加速電壓:19.000kv;延遲引出電壓:14.920kv;反射器電壓:20.000kv;透鏡電壓:7.000kv;頻率:1.000Hz;激光器能量:75-80%;累加次數:90次;負離子模式。
在MALDI電離方式下,基質可以吸收激光能量轉移給待測物使待測化合物電離,負離子模式下形成去質子峰,從而被質譜檢測。實驗證明,參考圖4,該基質對于以上氨基酸均具有較好的分析性能。圖中待測物的量均為500pmol,可以看出該利用基質分析靈敏度較高。除了圖中列出的化合物以外,對圖中每一類物質的同類化合物,該基質均可以得到較好的分析結果。
實施例3對絲氨酸以及苯丙氨酸的富集和檢測
采用含有六方氮化硼納米片的基質溶液對含有不同小分子的溶液進行固相微萃取以及質譜分析。
將絲氨酸(ser)和苯丙氨酸(phe)各自配制成濃度為5μmol/L的溶液,分別取495μL(與ser溶液混合)與5μL(與phe溶液混合)0.25mg/mL的h-BN納米片懸浮液混合。搖床中孵育15min后離心(10000g),將沉淀用體積比為1:1的甲醇水溶液重懸為10μL,取1μL點靶。進行質譜測試。質譜條件為:電壓:加速電壓:19.000kv;延遲引出電壓:14.920kv;反射器電壓:20.000kv;透鏡電壓:7.000kv;頻率:1.000Hz;激光器能量:75-80%;累加次數:90次;負離子模式。
參考圖5,分別為絲氨酸(ser)富集前(圖5A)、富集后(圖5B)以及苯丙氨酸(phe)富集前(圖5C)、富集后(圖5D)的MALDI-TOF MS質譜圖。從富集前后對比可見,待測物信號均得到了不同程度的增強。具有SPE吸附劑和MALDI基質雙重能力的六方氮化硼納米片具有在檢測低分子量物質的同時簡化樣品制備的潛力。進一步的工作可以應用到大氣,水體環境污染物的直接提取分析。
實施例4對水體污染物(硝基多環芳烴)的富集和檢測
將9-硝基蒽配制成系列濃度分別為50μmol/L、1μmol/L以及0.5μmol/L的溶液,溶劑為體積比為1:1的甲醇水溶液。分別取495μL上述濃度的9-硝基蒽溶液與5μL六方氮化硼納米片濃度為0.1mg/L的基質溶液混合。搖床中孵育15min后離心(10000g),將沉淀用體積比為1:1的甲醇水溶液重懸為10μL,取1μL點靶。進行質譜測試。質譜條件為:電壓:加速電壓:19.000kv;延遲引出電壓:14.920kv;反射器電壓:20.000kv;透鏡電壓:7.000kv;頻率:1.000Hz;激光器能量:75-80%;累加次數:90次;負離子模式。參考圖6(a),在負離子模式下,不對待測溶液進行富集時,50μmol/L的9-硝基蒽溶液是檢測不到的。
參考圖6(b)以及圖6(c)對濃度為1μmol/L以及0.5μmol/L的9-硝基蒽溶液經過富集之后,均可以檢測出9-硝基蒽。富集過程如下:取495μl濃度為1μmol/L或者0.5μmol/L 的9-硝基蒽溶液,與5μL實施例1中配置的基質溶液混合,超聲15分鐘之后,在搖床中孵育15min。在1000g條件下進行離心15分鐘,取離心后的沉淀,使用體積比為1:1的甲醇水溶液重懸為10μL,取1μL點靶。
因此,該基質可用于水體污染物(9-硝基蒽)的檢測。另外,還可對其進行定量檢測,線性范圍為1-50μmol/L。
實施例5微生物代謝物的分析
液體培養的大腸桿菌生長到OD=0.8~1.0后收集。收集的菌體用50倍pH值為7.4的磷酸鹽緩沖液洗滌后,懸浮的細菌用去離子水置換,9000g離心留沉淀,向10mg沉淀中加入3倍體積的體積分數為80%的甲醇溶液,懸浮后靜置10min,再經9000g離心后取上清液。取5μl上清液與495μl濃度為0.1mg/L的六方氮化硼納米片基質溶液進行混合,取1μl混合溶液點靶進行質譜檢測。質譜條件為:電壓:加速電壓:19.000kv;延遲引出電壓:14.920kv;反射器電壓:20.000kv;透鏡電壓:7.000kv;頻率:1.000Hz;激光器能量:75-80%;累加次數:90次;負離子模式。
參考圖7,在負離子模式下,可以產生豐富的大腸桿菌代謝物譜峰。并且標準培養(圖7a)與饑餓培養(圖7b)的大腸桿菌代謝物譜峰有明顯的差異。其中,質荷比為180.06的峰為酪氨酸,質荷比為267.31的峰為肌苷,質荷比在400-900之間的峰為脂類化合物。參考圖8,圖8a為圖7a中質荷比為400~900區域的放大圖。采用濃度同為0.25mg/L的鹽酸萘乙二胺溶液作為基質溶液,在相同的條件下(標準培養)對微生物代謝產物進行分析可以發現,采用鹽酸萘乙二胺溶液作為基質的質譜結果中(圖8b),檢測出的脂類化合物的特征峰明顯不如采用含有六方氮化硼納米片的基質溶液(圖8a)的質譜結果豐富。因此,含有六方氮化硼納米片的基質在質譜檢測中對于脂類物質的檢測具有選擇性。
實施例6鼠腦成像
取兩只小鼠做平行實驗,小鼠通過頸部脫臼法處死,冷凍的新鮮腦部在冷凍切片機上進行切片,切片厚度為10μm。隨后將切片貼于ITO玻璃片上,室溫真空條件下干燥30-60min,干燥之后,將含有0.1mg/L的六方氮化硼納米片基質溶液噴灑到組織切片上,進行質譜成像檢測。質譜條件:電壓:加速電壓:19.000kv;延遲引出電壓:14.920kv;反射器電壓:20.000kv;透鏡電壓:7.000kv;頻率:1.000Hz;激光器能量:75-80%;掃描分辨率為100μm,激光光斑為50μm;負離子模式。成像數據采用FlexImaging v3.0或者BioMap v3.8處理。
參考圖9可以看出,在小鼠腦檢測中,該基質最顯著的特點是傾向選擇性檢測硫苷脂,即位于質荷比為500~1000的峰。例如,位于888.6的C24:1(脂肪鏈含24個C,并包含一個不飽和鍵)硫苷脂。還檢測到N-乙酰基-L-天冬氨酸(N-Acetyl-L-aspartic acid),油酸(oleic acid),C18硫苷脂(C18sulfatide),C24:1硫苷脂(C24:1sulfatide)以及C24-OH硫苷脂(C24-OH sulfatide)。
此外,該基質也支持高分辨率的質譜成像。參考圖10,分別對兩只小鼠的切片進行檢測,對兩份切片樣品中質荷比(m/z)為888.8的硫苷脂(ST)(結構式見圖9)以及質荷比為(m/z)885.6的磷脂酰肌醇(PI)(結構式見式1)的含量進行檢測,由質譜成像圖中可以清晰地看出上述兩種物質在兩只小鼠腦切片中的位置分布以及含量(由圖中的亮度表示)。
在本說明書的描述中,參考術語“一個實施例”、“一些實施例”、“示例”、“具體示例”、或“一些示例”等的描述意指結合該實施例或示例描述的具體特征、結構、材料或者特點包含于本發明的至少一個實施例或示例中。在本說明書中,對上述術語的示意性表述不必須針對的是相同的實施例或示例。而且,描述的具體特征、結構、材料或者特點可以在任一個或多個實施例或示例中以合適的方式結合。此外,在不相互矛盾的情況下,本領域的技術人員可以將本說明書中描述的不同實施例或示例以及不同實施例或示例的特征進行結合和組合。
盡管上面已經示出和描述了本發明的實施例,可以理解的是,上述實施例是示例性的,不能理解為對本發明的限制,本領域的普通技術人員在本發明的范圍內可以對上述實施例進行變化、修改、替換和變型。
- 還沒有人留言評論。精彩留言會獲得點贊!