一種多孔鎳基合金電解析氫陰極材料的制備方法與流程
本發明涉及一種多孔鎳基合金材料的制備技術,特別是涉及一種ni-cr-fe-co電解析氫陰極材料的制備方法。
背景技術:
工業革命以來,傳統的化石燃料能源日益枯竭,化石燃料能源的儲藏量日益枯竭。及時開發清潔、價廉的新能源,以逐步取代現有的化石燃料,達到減少污染與溫室氣體排放、最終實現零排放的目的尤為重要。而氫能以其儲量豐富、熱值高、清潔、高效、便于運輸等特點在人們探索的眾多新能源中,引起人們越來越多的關注。氫能開發正在引發一場深刻的能源革命,并將成為21世紀理想的二次能源。目前常見的氫氣制備方法有化石燃料制氫、微生物制氫、光催化析氫、電解水制氫等。其中電解水制氫效率高,工藝過程簡單,幾乎不產生污染,應用比較廣泛。但由于目前的析氫電極具有較高的過電位和穩定性較差而導致能耗大,限制了其大規模生產。開發具有較高的析氫催化活性和電催化穩定性的新型電極材料很有必要。
過渡金屬ni是良好的析氫陰極材料。首先,過渡金屬ni的價電子層排布為3d84s2,含有未充滿的d電子層,原子h進入金屬中,電離出的電子與ni中未成對d電子形成了m-h鍵。通常過渡金屬中有未成對的d電子或空軌道,和氫反應都會生成m-h鍵。當金屬表面形成的吸附鍵鍵能遞增時,根據“火山型效應”,過渡金屬與h的反應速度先增大后減小。金屬ni具有適中的吸附h鍵能和較快的反應速率,從而表現出優異的電催化析氫反應性能。其次是因為它們大多都是非晶態合金,這種合金具有長程無序、短程有序的相結構。這種特別的相結構使得原子呈現出無規則且緊密的排列方式,這種結構明顯降低了析氫反應的活化能,從而展現出優異的電催化性能。由tafel公式可知,增大電極的比表面積可以提高電極的析氫催化活性,比如具有多孔結構的鎳鋁合金——雷尼鎳可以使鎳電極的表面積增大1000倍以上,于是可以得到相對較低的析氫過電位。如專利cn103103562a中的ni-co-w-cu-b多組分陰極材料包括基底和表面的電沉積ni-co-w-cu-b合金鍍層,其基底為具有更大表面積材料的銅網而非銅片,在同等條件下所得到的ni-co-w-cu-b多組分陰極材料產生了更好的產氫性能。可知,增大材料的表面積可以得到相對較低的析氫過電位,提高電極的析氫催化活性。如專利cn105483744a中也指出將鉻族金屬(cr、mo、w)添加到鐵系金屬(fe、co、ni)當中,可有效降低鐵系金屬表面析氫反應的活化能,從而提高其析氫催化活性。同時,鐵系金屬在堿性水溶液中的化學穩定性很高,自身不易腐蝕。如能合理選擇鉻族金屬與鐵系金屬所形成合金的元素配比,并控制合金的物相組成,便有希望獲得一種在堿性水溶液中析氫活化能低且耐腐蝕的合金催化劑。
本發明正是基于此而提出采用元素粉末反應合成法制備ni-cr-fe-co多孔鎳基電解析氫陰極材料。該材料具有較高的比表面積、較低的析氫過電位、相對優良的抗腐蝕性能、較好的化學穩定性和較高的機械強度等優點,制備工藝簡單環保,將對氫能源的開發與應用有很大的意義。
技術實現要素:
本發明目的在于提供一種具有豐富孔隙且孔徑相對較大的粉末冶金燒結多孔電解析氫陰極材料的制備方法。本發明采用ni、cr、fe、co粉制成孔隙豐富的多孔材料,利用其豐富的孔結構、較低的析氫過電位、優異的電催化活性和良好的化學穩定性,解決現有析氫電極催化活性不高、比表面積小、耐腐蝕性不好、抗斷電短路能力差和析氫不穩定等問題。
本發明公開了一種多孔鎳基合金電解析氫陰極材料的制備方法,其具體制備方法包括以下步驟:
(1)粉末配制:將ni、cr、fe、co四種高純元素粉末按一定質量百分比配好,其中cr、fe、co粉共占總質量的25~60%,余量為ni;
(2)粉末處理:將配制好的粉末放在v型混粉機上勻速混合8~16h后,加入粉末總質量1~3%的硬脂酸,再在40~60℃普通干燥箱中干燥6~12h;
(3)壓制成型:將混合均勻的粉料在50~200mpa的壓力下保壓30~120s后壓制成型,得到生坯;
(4)生坯燒結:將步驟(3)所制生坯置于真空燒結爐中進行燒結,真空度為1x10-2~10-3pa;燒結工藝為:保持5~15℃/min的升溫速度從室溫升至100~140℃,保溫15~40min;接著以3~10℃/min的升溫速度升溫至300~400℃,保溫40~80min;再以5~15℃/min的升溫速度升溫至550~650℃并在該溫度下保溫50~80min;然后以6~12℃/min的升溫速度升溫至850~950℃并在該溫度下保溫30~50min;隨爐冷卻至室溫,即得到所發明的多孔電解析氫陰極材料。
步驟(1)中所用ni粉、cr粉、fe粉與co粉的平均粉末粒徑為3~10μm。
步驟(1)中所用元素cr粉的質量百分比優選為20~42%。
步驟(1)中所用元素fe粉的質量百分比優選為4~12%。
步驟(1)中所用元素co粉的質量百分比優選為1~6%。
本發明采用上述技術方案的優點在于:
(1)開孔隙率高,比表面積大。本發明利用燒結過程中元素粉末之間的偏擴散效應使材料產生大量且分布均勻的孔隙,增大了材料的比表面積,降低了析氫過電位,提高了電極的析氫催化活性;
(2)協同催化,催化活性良好。本發明利用ni與cr、fe、co之間的催化協同作用使得ni-cr-fe-co合金在電解析氫過程中具有適中的吸附和脫附性能,降低了材料的析氫過電位,提高了電極的析氫催化活性;
(3)耐腐蝕性好,析氫穩定。本發明將cr添加到fe、co、ni中,使合金在堿性水溶液中的化學穩定性提高,自身不易腐蝕;
(4)工藝簡單環保,可批量生產。本發明中所用粉末ni、cr、fe、co來源廣泛,價格低廉,而且制備工藝簡單可控,生產過程中原材料利用率高,無污染產生,可實現工業化生產。
附圖說明
圖1為實施例1中制備的ni-cr-fe-co多孔電極的表面形貌圖。
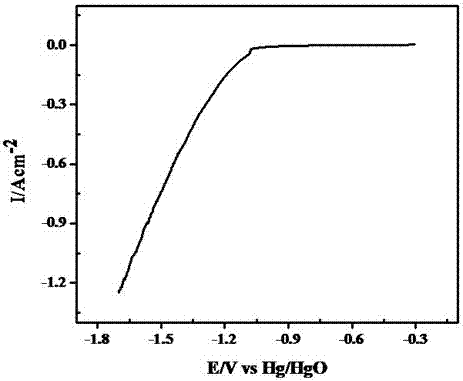
圖2為實施例1中制備的ni-cr-fe-co多孔電極的陰極極化曲線。
具體實施方式
下面結合具體實施例對本發明作進一步的描述。
實施例1
將ni、cr、fe、co四種高純元素粉末按質量百分比配好,其中cr含量為20%,粉末粒徑為3μm;fe含量為4%,粉末粒徑為3μm;co含量為6%,粉末粒徑為3μm;余量為粉末粒徑為10μm的ni粉。將配制好的粉末放在v型混粉機上勻速混合8h后加入粉末總質量1%的硬脂酸,再干燥6h,在冷壓機下以200mpa的壓力冷壓成形,保壓時間約為30s;將壓好的樣品置于真空爐中,在真空度為1x10-2pa的情況下保持5℃/min的升溫速度從室溫升至140℃,保溫40min;接著以10℃/min的升溫速度升溫至300℃,保溫80min;再以15℃/min的升溫速度升溫至650℃并在該溫度下保溫80min;然后以12℃/min的升溫速度升溫至950℃并在該溫度下保溫50min;隨爐冷卻至室溫,即得到所發明的多孔電解析氫陰極材料。
所得材料的微觀表面形貌圖如圖1所示,可見材料具有豐富的連通孔隙,且孔隙分布均勻,孔徑大小在1μm~12μm。
為了研究多孔鎳基合金析氫陰極材料的催化析氫性能,要將燒制的樣品用環氧樹脂密封并留出1cm2的表面積,在6mol/l的koh溶液中進行電化學測試。測試采用標準三電極體系,輔助電極為石墨,參比電極為hg/hgo,工作電極為燒結制得的ni-cr-fe-co樣品。測試所用的儀器為cs350電化學工作站,掃描速度為1mv·s-1,掃描范圍為0v~-2v,測試時采用恒溫水浴讓電解液保持在25℃。ni-cr-fe-co多孔電極的陰極極化曲線如圖2所示,當電極電位達到-1.5v時,電流密度為0.90a/cm2。
實施例2
將ni、cr、fe、co四種高純元素粉末按質量百分比配好,其中cr含量為42%,粉末粒徑為10μm;fe含量為8%,粉末粒徑為10μm;co含量為1%,粉末粒徑為3μm;余量為粉末粒徑為3μm的ni粉。將配制好的粉末放在v型混粉機上勻速混合10h后加入粉末總質量3%的硬脂酸,再干燥12h,在冷壓機下以50mpa的壓力冷壓成形,保壓時間約為120s;將壓好的樣品置于真空爐中,在真空度為1x10-3pa的情況下保持8℃/min的升溫速度從室溫升至100℃,保溫15min;接著以3℃/min的升溫速度升溫至300℃,保溫40min;再以5℃/min的升溫速度升溫至550℃并在該溫度下保溫50min;然后以6℃/min的升溫速度升溫至850℃并在該溫度下保溫30min;隨爐冷卻至室溫,即得到所發明的多孔電解析氫陰極材料。
重復實施例1中的制備樣品過程和電化學實驗步驟再進行電化學實驗,得到與實施例1相似的孔結構和電化學性能。
實施例3
將ni、cr、fe、co四種高純元素粉末按質量百分比配好,其中cr含量為30%,粉末粒徑為5μm;fe含量為10%,粉末粒徑為8μm;co含量為3%,粉末粒徑為6μm;余量為粉末粒徑為8μm的ni粉。將配制好的粉末放在v型混粉機上勻速混合16h后加入粉末總質量1.5%的硬脂酸,再干燥10h,在冷壓機下以180mpa的壓力冷壓成形,保壓時間約為90s;將壓好的樣品置于真空爐中,在真空度為5x10-3pa的情況下保持15℃/min的升溫速度從室溫升至120℃,保溫25min;接著以5℃/min的升溫速度升溫至400℃,保溫60min;再以8℃/min的升溫速度升溫至600℃并在該溫度下保溫70min;然后以8℃/min的升溫速度升溫至900℃并在該溫度下保溫40min;隨爐冷卻至室溫,即得到所發明的多孔電解析氫陰極材料。
重復實施例1中的制備樣品過程和電化學實驗步驟再進行電化學實驗,得到與實施例1相似的孔結構和電化學性能。
實施例4
將ni、cr、fe、co四種高純元素粉末按質量百分比配好,其中cr含量為25%,粉末粒徑為8μm;fe含量為12%,粉末粒徑為3μm;co含量為6%,粉末粒徑為10μm;余量為粉末粒徑為4μm的ni粉。將配制好的粉末放在v型混粉機上勻速混合12h后加入粉末總質量2%的硬脂酸,再干燥6h,在冷壓機下以120mpa的壓力冷壓成形,保壓時間約為30s;將壓好的樣品置于真空爐中,在真空度為2x10-3pa的情況下保持10℃/min的升溫速度從室溫升至140℃,保溫35min;接著以8℃/min的升溫速度升溫至340℃,保溫80min;再以12℃/min的升溫速度升溫至650℃并在該溫度下保溫60min;然后以10℃/min的升溫速度升溫至880℃并在該溫度下保溫50min;隨爐冷卻至室溫,即得到所發明的多孔電解析氫陰極材料。
重復實施例1中的制備樣品過程和電化學實驗步驟再進行電化學實驗,得到與實施例1相似的孔結構和電化學性能。
實施例5
將ni、cr、fe、co四種高純元素粉末按質量百分比配好,其中cr含量為35%,粉末粒徑為3μm;fe含量為6%,粉末粒徑為6μm;co含量為1%,粉末粒徑為4μm;余量為粉末粒徑為10μm的ni粉。將配制好的粉末放在v型混粉機上勻速混合16h后加入粉末總質量1%的硬脂酸,再干燥8h,在冷壓機下以50mpa的壓力冷壓成形,保壓時間約為100s;將壓好的樣品置于真空爐中,在真空度為8x10-3pa的情況下保持5℃/min的升溫速度從室溫升至130℃,保溫40min;接著以10℃/min的升溫速度升溫至300℃,保溫70min;再以10℃/min的升溫速度升溫至620℃并在該溫度下保溫80min;然后以6℃/min的升溫速度升溫至950℃并在該溫度下保溫40min;隨爐冷卻至室溫,即得到所發明的多孔電解析氫陰極材料。
重復實施例1中的制備樣品過程和電化學實驗步驟再進行電化學實驗,得到與實施例1相似的孔結構和電化學性能。
實施例6
將ni、cr、fe、co四種高純元素粉末按質量百分比配好,其中cr含量為30%,粉末粒徑為10μm;fe含量為4%,粉末粒徑為3μm;co含量為2%,粉末粒徑為8μm;余量為粉末粒徑為6μm的ni粉。將配制好的粉末放在v型混粉機上勻速混合8h后加入粉末總質量3%的硬脂酸,再干燥12h,在冷壓機下以200mpa的壓力冷壓成形,保壓時間約為80s;將壓好的樣品置于真空爐中,在真空度為6x10-3pa的情況下保持12℃/min的升溫速度從室溫升至100℃,保溫20min;接著以6℃/min的升溫速度升溫至360℃,保溫50min;再以15℃/min的升溫速度升溫至580℃并在該溫度下保溫50min;然后以12℃/min的升溫速度升溫至860℃并在該溫度下保溫30min;隨爐冷卻至室溫,即得到所發明的多孔電解析氫陰極材料。
重復實施例1中的制樣和電化學實驗步驟再進行電化學實驗,得到與實施例1相似的孔結構和電化學性能。
以上所述僅是對本發明的較佳實施方式而已,并非對本發明作任何形式上的限制,凡是依據本發明的技術實質對以上實施方式所做的任何簡單修改,等同變化與修飾,均屬于本發明技術方案的范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!