使碳納米材料的表面官能化的方法與流程
本申請要求2014年9月16日提交的新加坡專利申請No.10201405784X的優先權,對于所有目的其全部內容通過引用并入本文。
技術領域
本發明涉及一種采用空氣中的氧使碳納米材料的表面官能化的方法。
背景技術:
碳納米管(CNT)是準一維材料。在過去二十年中,由于碳納米管突出的機械、化學、電氣、電子和熱性能,人們對碳納米管進行了大量研究。它們的應用包括復合材料、傳感器、光伏器件、致動器、晶體管和能量存儲和轉換裝置。由于它們的高縱橫比(長度為微米級,直徑小于100nm)和強大的分子間范德華力,CNT通常形成高度纏結的束和網絡,使得它們難以在溶劑中分散。CNT的分散不均嚴重限制了它們的潛在應用。例如,對于衍生自基于CNT的懸浮液或納米復合材料的許多應用,其關鍵是獲得高水平的分散性。
已經探索了多種方法來分散CNT,包括共價和非共價改性。采用各種強酸的氧化反應通常是化學改性的常見且有效的出發點。酸氧化反應引入表面官能團,以獲得在水溶液中良好的分散性,且還能夠進一步改性,例如聚合物的接枝。許多強氧化劑已經被用于處理碳納米管,并且可以在CNT的表面上產生各種極性基團,包括羧基、羥基和酮基。這些極性基團可以與溶劑分子相互作用,從而極其有效地提高碳納米管在水溶液中的分散性。盡管酸/化學氧化反應是有效的,但這種方法的缺點是顯而易見的。首先,使用大量的強酸會導致污染,減少潛在的大規模工業水平的生產。純化工藝過于繁瑣耗時,通常包括多步洗滌、過濾、離心、滲析和干燥。此外,在CNT的酸氧化處理期間會消耗大量的水,并產生大量廢水。此外,由于損耗和難于控制,酸氧化反應通常得到較低的產率。CNT的濕化學處理通常需要冷凍干燥以確保再分散性,因此其能耗很高。
在碳納米管的表面上非共價包裹特定的聚合物是改善碳納米管的分散性的另一種方法。與氧化反應不同,物理包裹不會在碳納米管上產生任何新的基團。相反,聚合物鏈與碳納米管物理上相互作用,形成超分子結構。非共價聚合物包裹可以良好地保持碳納米管的結構。然而,許多那些有效的包裹聚合物必須被特別設計和合成。這個工藝也消耗大量的溶劑,使其難以實施。
因此,仍然需要提供一種使碳納米材料(例如碳納米管)官能化的方法,以在有機溶劑中有效分散。
技術實現要素:
表面官能化通常是實現碳納米材料(例如碳納米管)的良好分散性的先決條件,這對于它們的廣泛應用是至關重要的。本文中描述了通過空氣退火使碳納米管的表面官能化的簡易且有效的途徑。退火的碳納米管在有機溶劑中具有優異的分散性。新的途徑是清潔且環保的,幾乎零化學制品使用和零廢棄物產生。形態和化學結構的定性和定量分析顯示,納米管在空氣退火期間的表面氧化反應是明確的且可以非常好地進行控制。研究還顯示,空氣退火對經處理的納米管中的石墨結構造成的損壞很小。還討論了這種簡易官能化方法的機械作用方面。
根據本發明的各種實施例,公開了采用空氣中的氧使碳納米材料的表面官能化的方法。該方法包括采用空氣對碳納米材料進行退火。該方法還包括在退火期間向碳納米材料供應一種或多種稀釋氣體。一種或多種稀釋氣體對碳納米材料表面的官能化是惰性的。
優選地,一種或多種稀釋氣體包括氮氣。
附圖說明
在附圖中,相同的附圖標記在不同視圖中通常指代相同的部件。附圖不一定按比例繪制,而通常著重于說明各種實施例的原理。在下面的說明書中,參考以下的附圖描述本發明的各種實施例。
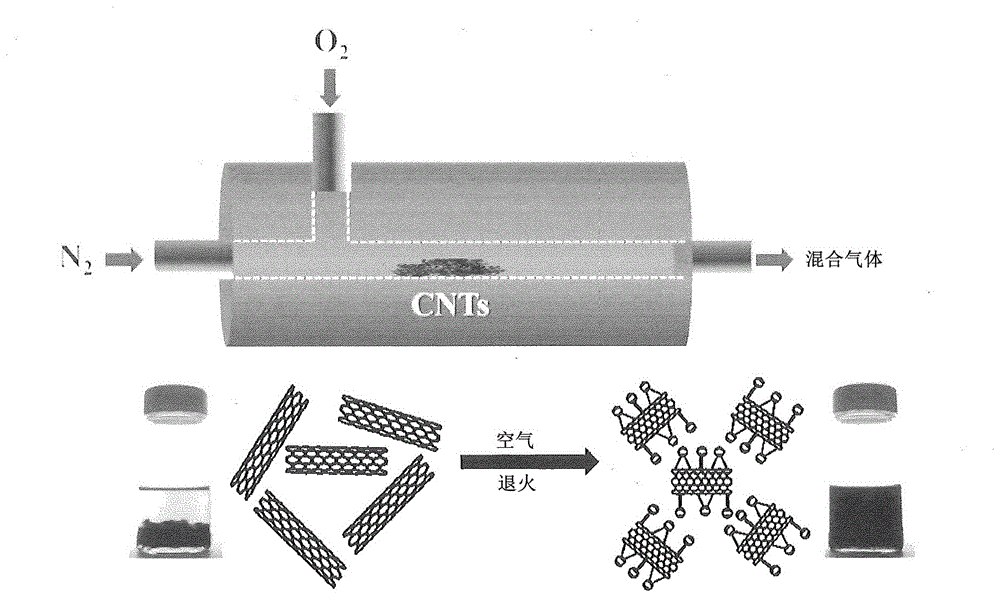
圖1示出了CNT的當前空氣退火工藝及其在處理前后在有機溶劑中的分散狀態的示意圖。
圖2示出了退火條件、樣品代碼和分散試驗。示出的百分比值表示退火后的質量殘留率或產率。將0.5mg CNT混合于10ml乙醇中,水浴超聲處理1小時,制備所有分散樣品。在靜止儲存1周后拍攝數碼圖像。(*由于過度過氧化導致的CNT的低產率)
圖3示出了(a)原始CNT,(b)混合酸處理的CNT,(c)在500℃退火30min的樣品B1和(D)在450℃下退火120min的樣品A4的TEM圖像。
圖4示出了(a)原始CNT,(b)B1(500℃,30min),(c)A4(450℃,120min),(d)酸處理的樣品和(e)在650℃下處理的樣品的拉曼光譜和ID/IG值。
圖5示出了含有0.2wt%(a,b)原始CNT和(c,d)樣品A4的PVDF/CNT復合材料的FESEM圖像。
圖6示出了(a)原始CNT,(b)在550℃下空氣退火30min的樣品C1和(c)酸處理的CNT的XPS光譜。
圖7示出了(a)原始CNT,(b)B1,(c)樣品C1和(d)酸處理的CNT的去卷積的高分辨率XPS C 1s光譜。它們相應的O 1s光譜分別在(e)、(f)、(g)和(h)中示出。
圖8示出了所提出的CNT表面通過空氣退火的官能化機理:(a)原始CNT,(b)空氣退火后過氧化物中間體的形成,(c)鍵重排和(d)官能化CNT。應注意到,I和II分別代表CNT樣品上的不同位點,顯示了分別通過1,4-和1,2-過氧化反應形成C-O-C和C=O的機理。
圖9示出了表1:根據空氣退火的CNT的XPS光譜檢測的不同官能團的組成。
圖10示出了在空氣條件下原始CNT的TGA曲線,CNT的分解可以大致分為四個階段。在500℃之前,CNT沒有明顯的變化。微小的重量損失是由于去除了水分和一些其它吸收的材料。在該溫度范圍內可能發生緩慢的氧化反應。在500℃至550℃之間,重量明顯減少,但分解速率相當緩慢。在550℃之前的高溫,有希望將CNT官能化而不發生嚴重的分解。在550℃和700℃之間,CNT迅速燃燒成灰;超過700℃時,僅剩下金屬氧化物。為了證實空氣退火的假設,在650℃處理CNT的試驗樣品。由于高的分解速率,將溫度升高到設定值并停止,沒有任何等溫退火過程。
圖11示出了在650℃下空氣退火的CNT試驗樣品的TEM圖像(A,B)。與原始CNT的相當整潔的表面相比,雖然截斷效應不明顯,但來自試驗樣品的CNT在高溫處理后看起來彼此粘連。從(B)中的高倍放大圖片可以看出,CNT壁的晶體結構嚴重損壞,并且大量碳質碳附著在CNT的內表面和外表面上。這表明在該溫度下空氣對CNT是非常有侵蝕性的。分散試驗顯示,在超聲處理停止后,原始CNT開始同時沉降。事實上,原始CNT形成絮狀結構且甚至在超聲處理下也不能均勻分散。相比之下,試驗樣品在光下可以穩定幾個月而沒有明顯的沉積物。
圖12示出了在各種溶劑中的原始CNT(左)和退火樣品(B1,右)的分散測試。將所有分散樣品超聲處理60min,并在儲存1周后拍攝數碼照片(0.5mg CNT在10ml溶劑中)。空氣退火的CNT容易分散在普通有機溶劑中,且懸浮液穩定超過一周,而原始CNT在從超聲波儀中取出瓶子后立即沉淀。
圖13示出了表2。根據酸處理的CNT的XPS光譜檢測的不同官能團的組成。
具體實施方式
以下詳細的說明書參考附圖,附圖通過圖解的方式示出了可以實踐本發明的具體細節和實施例。這些實施例具有充分詳細的描述,使本領域技術人員能夠實踐本發明。可以利用其他實施例且可以在不脫離本發明的范圍內進行改變。各種實施例不一定是相互排斥的,因為一些實施例可以與一個或多個其他實施例組合以形成新的實施例。
本文描述了通過在適度的高溫下采用空氣對碳納米材料進行退火來制備表面官能化的碳納米材料(例如碳納米管)的簡易方法。在這種情況下,空氣中的氧氣用作為有效的氧化劑以官能化碳納米材料。此外,通過調整退火溫度、持續時間、空氣和稀釋氣體的體積比以及相應的空氣和稀釋氣體流速,氧化反應是高度可控的。其結果是,在表面上生成有助于碳納米材料在有機溶劑中分散的官能團,同時非常好地維持碳納米材料的機械和結構完整性。這是一種幾乎不含化學制品的方法,不使用任何溶劑、酸或水。由于省去了對處理后的碳納米材料的冷凍干燥過程,極大地節省了能量和時間。試驗結果證實空氣退火的碳納米材料可以特別好地分散在有機溶劑中。詳細分析顯示,與酸處理的對應物相比,在有機溶劑中的這種意想不到的令人滿意的分散行為可以歸因于它們不同的表面官能團,即不存在羧基和羥基。此外,空氣退火對碳納米材料造成的結構損壞更少。
因此,本發明的各種實施例涉及采用空氣中的氧氣使碳納米材料的表面官能化的方法。
在本文中,碳納米材料包括單壁、雙壁或多壁碳納米管、碳納米纖維、石墨烯、活性炭、炭黑、洋蔥碳(carbon onion)或納米級金剛石和類金剛石(diamondoid)。根據各種實施例,本文中詳細描述了包含碳納米管的碳納米材料,作為示例性的實施例,并且應當理解,本文公開的范圍不限于碳納米管。
如本文所使用的,術語“碳納米管”(CNT)指圓柱形的單壁、雙壁或多壁結構,其中該結構的至少一個壁主要由碳組成。一般而言,碳納米管可以通過例如電弧放電法、激光燒蝕法和化學氣相沉積法形成。在形成之后,可以預處理或不處理碳納米管。
電弧放電法通過在通常填充有低壓惰性氣體的外殼中,電弧汽化首尾相連放置、空間間隔大約1mm的兩個碳棒來制造CNT。直流電在兩個電極之間產生高溫放電。放電使其中一個碳電極的表面汽化,并在另一個電極上形成小的棒狀碳原子沉積。
在激光燒蝕法中,可以通過在流動的氬氣中在高溫下用鈷和鎳的催化劑混合物對石墨棒進行激光汽化,然后在真空中熱處理以除去雜質來制備CNT。首個激光汽化脈沖后面可以跟隨著第二個脈沖,以更均勻地汽化目標。使用兩個連續的激光脈沖將沉積成煙灰的碳的量降至最少。第二個激光脈沖打碎由第一個激光脈沖燒蝕的較大顆粒,并將它們注入到生長的納米管結構中。通過改變生長溫度、催化劑組成和其它工藝參數,可以改變平均納米管直徑和尺寸分布。
化學氣相沉積法(CVD)也可以用于生產CNT。它可以通過分解由過渡金屬(例如鎳和鈷)催化的含碳分子而進行。在熱CVD中,含碳氣體混合物通過常規熱源加熱,例如電阻式或感應加熱器、火爐或IR燈。為了引起納米管生長,將工藝氣體(例如氨氣或氮氣)和含碳氣體(例如乙炔或甲烷)引入反應器中。納米管在金屬催化劑的位置上生長,從而使含碳氣體在催化劑顆粒的表面分裂,并且碳被轉移到顆粒的邊緣,在那里形成納米管。等離子體增強CVD(PECVD)通過在氣體混合物中應用點燃的放電來改進該方法。
在碳納米管中的外殼的數量可以從一個(即構成單壁碳納米管(SWNT或SWCNT))變化至多達50個外殼,在這種情況下,其被稱為多壁碳納米管(MWNT或MWCNT)。在這種結構中的每對相鄰的外殼在層之間具有大約~0.34納米級的間距,其中外殼可以是同軸的。可用于本發明的碳納米管的實例包括但不限于單壁碳納米管、雙壁碳納米管(DWNT或DWCNT)、多壁碳納米管、碳納米管束及其任意組合。在一些例舉的實施例中,碳納米管是單壁碳納米管。
碳納米管可以是金屬碳納米管,或半導體碳納米管,或兩者的組合。碳納米管可以具有任何長度和直徑。每個碳納米管可以具有大約0.3至200nm的直徑,例如大約3至200nm,大約1至100nm,大約0.3至50nm,或大約1至5nm。在一些實施例中,每個碳納米管可以具有大約0.5至300μm的長度,例如大約0.5至200μm,大約0.5至100μm或大約0.5至50μm。碳納米管的直徑通常為0.3至50納米,且具有的長度為0.5至100微米。原子力顯微鏡(AFM)和/或拉曼散射光譜可以例如用于檢測單壁碳納米管的尺寸規格。一般而言,碳納米管的長度越長,納米管纏結的可能越大。其結果是,可以形成碳納米管的纏結塊或簇。
在本文中,表面官能化和相關術語通常指對碳納米材料的表面進行改性。具體地,退火后,在碳納米材料的表面上形成官能團,例如醚基(C-O-C)和醌基(C=O)。甚至更具體地,在碳納米材料的表面上不會形成極性官能團,例如羥基(-OH)和羧基(-COOH)。
該方法包括采用空氣對碳納米材料進行退火。一般而言,例如,退火指通過加熱至預定溫度,保持一定時間,然后冷卻至室溫來處理碳納米材料。在一個實施例中,碳納米材料可以在退火設備中退火,例如商業產品熱重分析儀(Q 500,TA儀器(TA Instruments))。其它合適的退火設備包括管式爐、間歇式加熱爐或具有可控制溫度的機構的任何容器、設備或裝置。
氧化劑,即空氣中的氧氣,可以在退火期間以恒定的流速供應給碳納米材料。例如,可以以1ml/min、2ml/min、3ml/min、4ml/min、5ml/min、6ml/min、7ml/min,8ml/min、9ml/min、10ml/min或更大的流速向退火設備中的碳納米材料供應空氣。
在一個公開的實施例中,以5ml/min的恒定流速供應空氣。
空氣也可以以可變的流速(例如以增加的速率或減小的速率)供應至碳納米材料。
該方法還包括在退火期間向碳納米材料供應一種或多種稀釋氣體。一種或多種稀釋氣體對碳納米材料表面的官能化是惰性的。換句話說,一種或多種稀釋氣體用于稀釋在退火期間接觸碳納米材料的氧氣(或空氣)的濃度(v/v)。一種或多種稀釋氣體在改性碳納米材料的表面中沒有作用或具有極微小的作用,即對碳納米材料的官能化是化學惰性的。
在退火期間,接觸碳納米材料的空氣的濃度可以通過改變空氣與一種或多種稀釋氣體的體積比來控制。因此,在各種實施例中,空氣與一種或多種稀釋氣體的體積比可以在1:2和1:20之間,例如1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14、1:15、1:16、1:17、1:18、1:19或1:20。
在一個公開的實施例中,空氣與一種或多種稀釋氣體的體積比可以固定為1:10。這可以例如通過操作在退火期間供應到碳納米材料的一種或多種稀釋氣體的流速來實現。在一個示例性實施例中,對于固定的空氣流速5ml/min,一種或多種稀釋氣體的流速可以固定為50ml/min。如在前面的段落中提到的,其它體積比,和空氣與一種或多種稀釋氣體的其它流速比也是合適的。
如在前面的段落中提到的,任何稀釋氣體均適合用于本方法,只要該氣體能用于稀釋在退火期間供應的空氣的濃度,且該氣體不參與碳納米材料表面的官能化。
在各種實施例中,一種或多種稀釋氣體包括但不限于氮氣、氦氣、氬氣、氪氣、氙氣、二氧化碳和氡氣。
在一個公開的實施例中,稀釋氣體由氮氣組成。
在另一個實施例中,稀釋氣體包括氮氣和一種其它稀釋氣體的混合物。
在各種實施例中,碳納米材料可以在450℃和550℃之間的退火溫度下退火。在碳納米材料是碳納米管的情況下,已經發現在低于450℃的溫度下的退火不足以使表面充分官能化,而在高于550℃的溫度下退火更長的時間,會導致過多的重量損失,因此導致較低的產率。在實施例部分列舉了更多關于退火溫度的適合性的討論,且在圖2中進行了圖解。
在各種實施例中,碳納米材料可以在5℃/min或更高的加熱速率下退火。例如,碳納米材料可以在5℃/min、6℃/min、7℃/min、8℃/min、9℃/min、10℃/min、11℃/min、12℃/min、13℃/min、14℃/min、15℃/min、16℃/min、17℃/min、18℃/min、19℃/min或20℃/min的加熱速率下退火。
在一個公開的實施例中,碳納米材料可以在5℃/min的加熱速率下退火。
已經發現,碳納米材料的退火時間可能對碳納米材料中的氧含量的產量和質量有影響。因此,這會影響官能化的碳納米材料在有機溶劑(例如但不限于乙醇、丙酮、氯仿和N,N-二甲基甲酰胺)中的分散性。有關此主題的更多討論可以在實施例部分中找到。
因此,在各種實施例中,將碳納米材料退火150min或更少。例如,退火時間可以是30min、35min、40min、45min、50min、55min、60min、65min、70min、75min、80min、85min、90min、95min、100min、105min、110min、115min、120min、125min、130min、135min、140min、145min或150min。
總之,本文描述了通過空氣退火將碳納米材料(例如碳納米管)的表面官能化的簡易且有效的途徑。退火的碳納米管在有機溶劑中具有優異的分散性。新的途徑是清潔且環保的,幾乎零化學制品使用和零廢棄物產生。形態和化學結構的定性和定量分析顯示,納米管在空氣退火期間的表面氧化反應是明確的且可以非常好地進行控制。研究還顯示,空氣退火對經處理的納米管中的石墨結構造成的損壞更小。這個簡易的官能化方法的機械作用方面也在實施例部分中討論。
為了使本發明易于理解并實現實際效果,現在將通過下面的非限制性實施例描述具體的實施例。
實施例
實驗
材料
平均直徑為10nm和平均長度為1.5μm的多壁碳納米管購自比利時的Nanocyl公司。表面積為250-300m2/g。聚(偏氟乙烯)(PVDF)粉末購自Alfa Aesar。使用的溶劑,包括分析級乙醇、丙酮、氯仿和N,N-二甲基甲酰胺(DMF)購自Sigma Aldrich。
空氣退火
退火工藝在熱重分析儀(Q 500,TA Instruments)中進行。將未經任何預處理的原始CNT放置在鉑舟中,并在5ml/min的空氣流速下,以5℃/min的速率加熱至450℃和550℃之間的預設溫度。使用瞬時流量為50ml/min的N2,因為初步試驗表明需要稀釋以實現CNT的受控的表面氧化反應。該工藝的示意圖在圖1中示出。為了比較,還制備和研究了酸處理的MWCNT。
表征
采用高分辨率透射電子顯微鏡(TEM,Carl Zeiss Libra 120Plus)研究CNT的形態。采用場發射掃描電子顯微鏡(FESEM)研究制備的復合材料樣品的形態。通過首先將樣品浸入液氮中以制備冷凍斷裂的表面,接著采用鉑涂覆45s制備SEM樣品,然后利用JEOL JSM-7600F觀察。采用配備有軸超光譜儀(Axis Ultra spectrometer)(Kratos Analytical)的X射線光電子能譜(XPS)進行碳納米管的組分分析。采用在15kV下操作的單色Al KαX射線(1486.7eV)用作為光源。采用以氬離子激光器(488nm,20mW)作為激發源的Witec alpha300SR光譜儀記錄拉曼光譜。對于定量分析,在每個樣品的差異位置記錄五個光譜。
制備酸處理的CNT用于比較研究
制備酸處理的CNT用于比較研究,制造工藝如下。在第一步中,將1.0g原樣MWCNT轉移至裝有200ml水的透明瓶中,以進行探頭超聲波處理30min(3s開和3s關,100%振幅)。超聲處理后,采用0.2μm膜過濾渾濁溶液。產物在真空干燥箱中干燥過夜以排出水分。將從步驟1獲得的產物轉移到含有75ml硫酸和25ml硝酸(v/v=3:1)的250ml圓底燒瓶中。將溶液在水浴超聲波儀中在室溫下超聲處理30min,以將CNT分散在混合酸中。接著,將MWCNT和混合酸在劇烈磁力攪拌下在90℃下回流60min。然后,采用孔徑為0.2μm的耐酸性膜過濾稀釋的溶液。采用去離子水進行多次洗滌,直到官能化的CNT的pH為大約6至7為止。然后采用丙酮沖洗過濾后的CNT以除去大部分洗滌水。將產物在真空干燥箱中在50℃下減壓干燥過夜,然后獲得最終樣品。
空氣退火的CNT在聚合物中的分散性
采用大功率超聲處理(超聲處理器VCX 130,SONICS)在DMF中處理0.2wt%CNT 5min。然后,將溶解在50ml DMF中的PVDF與CNT溶液通過磁力攪拌1h和水浴超聲處理1h進行混合。將均一的PVDF/CNT/DMF溶液在100℃和120℃下在真空干燥箱中干燥過夜。采用高分辨率場發射SEM檢測冷凍斷裂的表面。
結果與討論
本發明的動機之一是在高溫下采用空氣中的氧氣有效地官能化碳納米管,而不損壞它們的完整性,且還得到最高的可能的產率。因此,在本研究中,空氣中的氧氣濃度有意采用高達10倍的N2進一步稀釋,以確保該工藝被良好地控制。
基于TGA分析(圖10)和初步研究,將合適的溫度范圍設定在450℃和550℃之間,以進行詳細研究。事實上,不難理解,氧化反應只能在臨界溫度以上發生,且隨著溫度升高而加速,太高的溫度會導致CNT在短時間內過度氧化,導致過多的重量損失并因此導致產率低。例如,在650℃處理的CNT導致59%的重量損失,即低的產率。雖然處理后的納米管可以形成幾個月內都穩定的溶劑分散體,但采用TEM(圖11)和拉曼光譜的更詳細的研究顯示石墨結構遭到實質損壞。圖2示出了退火的CNT的分散狀態。還示出了樣品代碼、它們相應的退火溫度/時間和百分比產率。
注意,一般正如人們可預期的,處理后的CNT的分散性隨著退火溫度和時間得到提高。在低于450℃的溫度下退火不足以產生充分的表面官能化,而在高于550℃的溫度下退火更長的時間會引起過多的重量損失,因此導致較低的產率。圖2清楚地表明,為了實現CNT在有機溶劑中的良好分散性,同時保持高產率以及避免由于過度氧化而造成的結構損壞(參見后面段落中關于拉曼光譜的討論),控制退火條件是至關重要的。樣品A4和B1由于同時具有高產率和優異分散性而突出。為了進一步理解空氣退火對CNT的官能化的作用,對處理后的CNT進行詳細的定性和定量分析。
圖3示出了空氣退火的CNT(樣品B1,A4)的TEM圖像。還分析了原始CNT和酸處理的CNT用于比較。可以清楚地看出,與原始CNT相比,除了在CNT表面上有微小的變化(參見圖3的(c)和(d)),樣品A4和B1沒有顯示出顯著不同的整體形態。這些樣品的重量損失都小于15%。這些結果證明,這種簡易的空氣退火可以很好地用于CNT處理,以相對高的產率實現優異的分散性,而不會對石墨結構的完整性造成過度的損壞。然而,在酸處理的樣品中,觀察到相對厚的無定形層(參見圖3的(b)),表明在CNT的石墨結構中具有更嚴重的損壞。酸處理的CNT所報告的這種無定形層對它們的熱穩定性和電性能具有不利影響。因此,對于CNT處理,控制空氣退火在保持固有的CNT結構方面明顯具有優勢。
已知,CNT的拉曼光譜對任何化學改性都是敏感的。在1360cm-1附近的所謂D帶與有序度密切相關,而在1585cm-1處的G帶源自石墨碳。D帶的強度隨著缺陷位置、雜質和無定形碳的量的增加而增加。ID/IG強度比是含CNT的碳材料上的缺陷的常用指示,因為較高的ID/IG比值表示樣品中更多的缺陷。圖4比較了原始CNT和空氣退火的CNT的拉曼光譜。圖4中示出的拉曼光譜分析顯示,樣品A4和B1中的ID/IG比值與未處理的原始CNT中的ID/IG比值非常接近。實際上,樣品B1的ID/IG比值略低于原始CNT的ID/IG比值(0.91比0.94),這可能是由于在500℃退火期間,選擇性去除CNT中的無定形碳雜質的原因。另一方面,在650℃處理的樣品和酸處理的CNT的ID/IG比值明顯較高,分別為1.15和1.12,這意味著在兩個樣品中存在更嚴重的結構損壞。實際上,酸處理的CNT的拉曼光譜顯示出明顯的光譜帶偏移,這很可能是由于在酸處理期間,CNT表面上形成無定形層。這再次顯示了,在受控條件下通過空氣退火的CNT的官能化在引起結構損壞上侵入性更小的優點。
酸處理的CNT通常在水性介質和某些高極性溶劑中分散良好,與酸處理的CNT不同,空氣退火的樣品A4和B1可以分散在幾種極性有機溶劑中。除了在乙醇中,退火樣品在包括丙酮、氯仿和DMF的常見有機溶劑中顯示出優異的分散行為(參見圖12)。應當指出的是,為了保持CNT的固有性質和結構完整性,處理后的CNT(具有很少或沒有結構損壞)在有機溶劑中的這種優異的分散性是非常令人滿意的特征。這對于CNT的應用特別重要,例如作為導電涂層或電極。
此外,在有機溶劑中的良好分散性是CNT通過簡易的空氣退火處理可以容易地分散在聚合物基體中的指示。為了說明CNT/聚合物復合材料的這種潛力,將空氣退火的CNT的分散性與PVDF基體中的原始CNT進行比較。圖5比較了使用相同的混合方法制備的PVDF中的空氣退火的CNT樣品(樣品A4)和原始CNT(均為0.2wt%)的FE-SEM圖像。可以看出,原始CNT在聚合物基體中形成明顯且大的聚集體(參見圖5的(a)和(b)),而樣品A4的納米管單獨地分散并均勻分布在整個聚合物基體中(參見圖5的(c)和(d))。到目前為止,在聚合物基體中實現CNT的這種優異的分散性而不進行冗長和費力的處理程序是困難的,其大規模實施是困難的且成本高。因此,我們認為通過空氣退火來官能化的這種簡易方法應該是非常有意義的且對CNT的各種應用是有用的。
XPS是研究CNT上的元素組成和官能團的有用工具。為了理解表面官能化的詳細機理,均采用XPS進行空氣退火的CNT的定性和定量分析。圖6比較了原始CNT、空氣退火的CNT和酸處理的CNT的C 1s和O 1s XPS光譜。由于雜質或催化劑殘留,在原始CNT中發現了氧含量是。在550℃下劇烈退火30min的樣品(C1)中的氧含量明顯增加。然而,氧含量仍低于酸處理的CNT中的氧含量。XPS觀察結果與前面討論的TEM和拉曼分析一致。這再次表明空氣退火可以是CNT的可控且侵入性較小的表面官能化。重要的是注意到,與原始CNT相比,僅在450℃及以上處理的樣品中觀察到氧含量的明顯增加。一系列所選樣品的總氧含量概括在圖9的表1最后一欄中。樣品A1、B1、A4和C1的氧含量分別為5.56%、7.37%、7.42%和12.93%(C1),正如預期的,在較高溫度/較長時間顯示出逐漸增加的趨勢。
關于官能團的更詳細的信息通過XPS光譜的進一步定量分析獲得。在圖7中示出去卷積的C 1s和O 1s光譜。原始CNT和空氣退火的CNT的C 1s峰可以擬合成兩個子峰。結合能為284.8eV處的主峰對應于CNT中的石墨結構的sp2雜化碳原子,而在285.8eV處的次峰是指與氧鍵合的sp3原子。對于原始CNT和空氣退火的樣品A1、A4、B1和C1(圖9,表1,第一欄),sp2雜化碳含量分別計算為84.71%、82.52%、81.41%、80.25%和76.25%。退火樣品中的sp2碳含量僅有少量減少。然而,相比之下,測得的酸處理的CNT中的相應石墨sp2雜化碳含量僅為54.11%(圖14,表2),其遠遠低于任何一個空氣退火的CNT的sp2碳含量。這量化了石墨結構在酸處理期間的損壞程度遠遠大于空氣退火。相應地,O 1s光譜也可以擬合成兩個峰。531.9eV的較低結合能量峰歸屬于醌基(C=O),在533.2eV處的另一個峰歸屬于醚基(C-O-C)。仔細檢查圖9的表1中總結的官能團含量,其進一步顯示,增加退火溫度似乎比增加退火時間對C-O-C和C=O官能團的含量具有更顯著的影響(圖9,表1C 1s)。來自圖9的表1中的O 1s光譜的數據顯示,當在450℃、500℃和550℃下退火時,-O-(與C-O-C相關)和=O(與C=O相關)的相對含量是非常不同的。重要的是注意到,較高的退火溫度明顯有利于形成醚基(C-O-C),但會減少醌基(C=O)。例如,在450℃下處理的樣品A1中的官能團總量中的C-O-C基團含量為16.40%。在550℃下處理的樣品C1中,其增加至45.25%。還注意到,樣品B1(30min@500℃)和A1(120min@450℃)具有幾乎相同的總氧含量,即7.37%和7.42%。然而,樣品B1中的C-O-C與C=O比值為0.30,而A1中的C-O-C與C=O的比值為0.23。這些觀察結果表明,較短時間/較高溫度退火比較長時間/較低溫度退火生成更多的C-O-C基,而后者(即較長時間/較低溫度退火)產生更多的C=O基。
為了比較,圖7的(d)和(h)示出了酸處理的CNT的C 1s和O 1s光譜分別去卷積為四種不同的碳態和三種氧態。這表明與空氣退火相比,酸氧化反應導致非常不同類型的官能團。除了在284.8eV處的石墨sp2的主峰之外,在285.6eV、286.6eV和289.2eV處的峰表示存在C-OH、C-O-C和COOH,其中這兩個在空氣退火樣品中不存在。在531.9eV處的主要氧峰來自COOH,而在533.2eV處的峰來自醚基和羥基。然而,值得強調的是,在任何一個空氣退火的CNT中沒有發現羥基或羧基。我們認為,這兩個基團的缺失是解釋為什么空氣退火的CNT可以分散在有機溶劑而不是水溶液中的重要原因。
基于XPS分析,本文提出了在空氣退火期間在CNT上形成表面醚基和醌基的可能的反應機理。圖8示出了提出的機理。為了形成C-O-C基,第一個和速率控制步驟是1,4過氧化反應。接著,在兩個相鄰的C=C鍵中的π電子重新排列并同時形成連接至兩個氧原子的單鍵,而如圖8中的路線I所示,-O-O-鍵均勻斷裂。
另一方面,C=O基的形成可能開始于在石墨環上的1,2過氧化反應,且隨后是C-C鍵和O-O鍵的同時均裂,形成兩個醌基(圖8中的路線II)。在XPS分析中可以看出,較低溫度對產生C-O-C基不太有利,這意味著低溫不太有利于1,4過氧化反應。這可以合理地預期,平面石墨環中的1,4過氧化反應的活化能高于1,2過氧化反應的活化能。
結論
發現空氣退火是CNT的表面官能化的有效途徑。這是一種簡易的方法,其使得處理后的CNT具有高達90%的高產率。醚基C-O-C和醌基C=O的表面官能化都是均一的,使得處理后的CNT在包括乙醇、丙酮、氯仿和DMF的有機溶劑中具有優異的分散性。基于詳細的XPS光譜分析提出了合理的反應機理。已經發現空氣退火處理對CNT產生最小的損壞,這從拉曼和TEM分析都可以看出。實際上,XPS數據還定量地證實,空氣退火的CNT在sp2石墨碳結構中具有非常少的減少,并且仍可以均勻地分散在不同的有機溶劑中。定量結果表明,與酸處理相比,其具有在空氣退火期間較少結構損壞的明顯優點。因此堅定地認為這種簡易且環保的官能化方法具有可擴大的可能性,且其在涉及CNT的許多不同應用中是有用的。
“包括(comprising)”表示包括但不限于跟在詞語“包括”后的任何內容。因此,使用術語“包括”表明所列出的元件是必需的或強制性的,但是其它元件是可選的且可以存在或可以不存在。
“由……組成(consisting of)”表示包括且限于跟在短語“由……組成”后的任何內容。因此,短語“由……組成”表明所列元素是必需的或強制性的,且不存在其它元素。
本文示例性描述的本發明可以在沒有本文未具體公開的任何一個或多個元件,一個限制或多個限制的情況下適當地實施。因此,例如,術語“包括(comprising)”、“包含(including)”、“含有(containing)”等將被理解為廣泛地且無限制地。另外,本文中采用的術語和表達用作描述性而不是限制性術語,且在使用這些術語和表達時不打算排除所示的和所描述的特征或其部分的任何等同物,而是認識到,在所要求保護的本發明的范圍內的各種修改是可能的。因此,應當理解的是,雖然本發明已經通過優選實施例和可選特征具體公開,但是本領域技術人員可以進行本文公開的本發明的修改和變型,并且這樣的修改和變型被認為在本發明的范圍內。
關于與給定數值有關的“大約”,例如對于溫度和時間段,“大約”指包括在指定值的10%內的數值。
本文已廣泛且一般性地描述了本發明。屬于一般公開內容中的每個較窄種類和亞類組群也形成本發明的一部分。這包括本發明的一般性描述,其具有從該類除去任何主題的附帶條件或否定限制,而不管所排除的材料是否在本文中具體描述。
其它實施例在以下權利要求和非限制性實施例內。此外,在本發明的特征或方面根據馬庫什組描述的情況下,本領域技術人員將認識到,本發明也因此根據馬庫什組的任何單個成員或亞組來描述。
參考文獻
1.Baughman,R.H.;Zakhidov,A.A.;de Heer,W.A.,Carbon nanotubes-the route toward applications.Science 2002,297(5582),787-792.
2.Bachtold,A.;Hadley,P.;Nakanishi,T.;Dekker,C.,Logic circuits with carbon nanotube transistors.Science 2001,294(5545),1317-1320.
3.Ren,S.;Bemardi,M.;Lunt,R.R.;Bulovic,V.;Grossman,J.C.;Gradecak,S.,Toward efficient carbon nanotube/p3ht solar cells:active layer morphology,electrical,and optical properties.Nano letters 2011,11(12),5316-5321.
4.Qin,S.;Qin,D.;Ford,W.T.;Herrera,J.E.;Resasco,D.E.;Bachilo,S.M.;Weisman,R.B.,Solubilization and purification of single-wall carbon nanotubes in water by in situ radical polymerization of sodium 4-styrenesulfonate.Macromolecules 2004,37(11),3965-3967.
5.Spitalsky,Z.;Tasis,D.;Papagelis,K.;Galiotis,C.,Carbon nanotube-polymer composites:chemistry,processing,mechanical and electrical properties.Progress in Polymer Science 2010,35(3),357-401.
6.Mazov,I.;Kuznetsov,V.L.;Simonova,I.A.;Stadnichenko,A.I.;Ishchenko,A.V.;Romanenko,A.I.;Tkachev,E.N.;Anikeeva,O.B.,Oxidation behavior of multiwall carbon nanotubes with different diameters and morphology.Applied Surface Science 2012,258(17),6272-6280.
7.Chen,J.;Liu,H.;Weimer,W.A.;Halls,M.D.;Waldeck,D.H.;Walker,G.C.,Noncovalent engineering of carbon nanotube surfaces by rigid,functional conjugated polymers.Journal of the American Chemical Society 2002,124(31),9034-9035.
8.Star,A.;Stoddart,J.F.;Steuerman,D.;Diehl,M.;Boukai,A.;Wong,E.W.;Yang,X.;Chung,S.W.;Choi,H.;Heath,J.R.,Preparation and properties of polymer-wrapped single-walled carbon nanotubes.Angewandte Chemie International Edition 2001,40(9),1721-1725.
9.Shin,Y.-R.;Jeon,I.-Y.;Baek,J.-B.,Stability of multi-walled carbon nanotubes in commonly used acidic media.Carbon 2012,50(4),1465-1476.
10.Pimenta,M.;Dresselhaus,G.;Dresselhaus,M.S.;Cancado,L.;Jorio,A.;Saito,R.,Studying disorder in graphite-based systems by Raman spectroscopy.Physical Chemistry Chemical Physics 2007,9(11),1276-1290.
11.Li,C.;Wang,D.;Liang,T.;Wang,X.;Wu,J.;Hu,X.;Liang,J.,Oxidation of multiwalled carbon nanotubes by air:benefits for electric double layer capacitors.Powder technology 2004,142(2),175-179.
12.Moonoosawmy,K.R.;Kruse,P.,Ambiguity in the Characterization of Chemically Modified Single-Walled Carbon Nanotubes:A Raman and Ultraviolet-Visible-Near-Infrared Study.The Journal of Physical Chemistry C2009,113(13),5133-5140.
13.Osswald,S.;Flahaut,E.;Ye,H.;Gogotsi,Y.,Elimination of D-band in Raman spectra of double-wall carbon nanotubes by oxidation.Chemical Physics Letters 2005,402(4),422-427.
14.Roy,S.;Das,T.;Ming,Y.;Chen,X.;Yue,C.Y.;Hu,X.,Specific functionalization and polymer grafting on multiwalled carbon nanotubes to fabricate advanced nylon 12composites.Journal of Materials Chemistry A 2014,2(11),3961-3970.
15.Lee,H.-J.;Oh,S.-J.;Choi,J.-Y.;Kim,J.W.;Han,J.;Tan,L.-S.;Baek,J.-B.,In situ synthesis of poly(ethylene terephthalate)(PET)in ethylene glycol containing terephthalic acid and functionalized multiwalled carbon nanotubes(MWNTs)as an approach to MWNT/PET nanocomposites.Chemistry of materials 2005,17(20),5057-5064.
16.Roy,S.;Das,T.;Yue,C.Y.;Hu,X.,Improved Polymer Encapsulation on Multiwalled Carbon Nanotubes by Selective Plasma Induced Controlled Polymer Grafting.ACS applied materials&interfaces 2013,6(1),664-670.
17.Tang,X.-Z.;Cao,Z.;Zhang,H.-B.;Liu,J.;Yu,Z.-Z.,Growth of silver nanocrystals on graphene by simultaneous reduction of graphene oxide and silver ions with a rapid and efficient one-step approach.Chem.Commun.2011,47(11),3084-3086.
18.Tang,X.-Z.;Li,X.;Cao,Z.;Yang,J.;Wang,H.;Pu,X.;Yu,Z.-Z.,Synthesis of graphene decorated with silver nanoparticles by simultaneous reduction of graphene oxide and silver ions with glucose.Carbon 2013,59,93-99.
19.Compton,O.C.;Dikin,D.A.;Putz,K.W.;Brinson,L.C.;Nguyen,S.T.,Electrically conductive“alkylated”graphene paper via chemical reduction of amine-functionalized graphene oxide paper.Advanced Materials 2010,22(8),892-896.
20.Luo,Z.;Lim,S.;Tian,Z.;Shang,J.;Lai,L.;MacDonald,B.;Fu,C.;Shen,Z.;Yu,T.;Lin,J.,Pyridinic N doped graphene:synthesis,electronic structure,and electrocatalytic property.Journal of Materials Chemistry 2011,21(22),8038-8044.
21.Zhang,L.;Ji,L.;Glans,P.-A.;Zhang,Y.;Zhu,J.;Guo,J.,Electronic structure and chemical bonding of a graphene oxide-sulfur nanocomposite for use in superior performance lithium-sulfur cells.Physical Chemistry Chemical Physics 2012,14(39),13670-13675.
22.Biniak,S.;G.;Siedlewski,J.;A.,The characterization of activated carbons with oxygen and nitrogen surface groups.Carbon 1997,35(12),1799-1810.
23.Haubner,K.;Murawski,J.;Olk,P.;Eng,L.M.;Ziegler,C.;Adolphi,B.;Jaehne,E.,The route to functional graphene oxide.ChemPhysChem 2010,11(10),2131-2139.
24.Soo-Jin Park;Ki-Seok Kim,Surface characterization of carbon materials by X-ray photoelectron spectroscopy.Microscopy:Science,Technology,Application and Education2010,1905-1916.
25.Nikolay Dementev;Sebastian Osswald;Yury Gogotsi;Eric Borguet,Purification of carbon nanotubes by dynamic oxidation in air.Journal of Materials Chemistry 2009,19,7904-7908.
26.K.Behler;S.Osswald;H.Ye;S.Dimovski;Y.Gogosti,Effect of thermal treatment on the structure of multi-walled carbon nanotubes.Journal of Nanoparticle Research 2006,8,615-625.
- 還沒有人留言評論。精彩留言會獲得點贊!