一種雙金屬催化劑及其制備方法和應用與流程
本發明屬于化工,具體涉及一種雙金屬催化劑及其制備方法和應用,所述雙金屬催化劑為含有銀、鎳、鋅、銅、鉻、錳、鈷、鐵或鋯的鈀碳催化劑。
背景技術:
1、2,3-二氯吡啶和2,3,5-三氯吡啶是非常重要的化工中間體,被廣泛應用于農藥和醫藥等領域。
2、2,3-二氯吡啶是合成新型殺蟲劑氯蟲苯甲酰胺的重要物質。目前,2,3-二氯吡啶的合成方法有以下幾種:2,3,6-三氯吡啶還原法、2-氯吡啶合成法、3-氯吡啶合成法和煙酰胺為原料的合成方法。其中,2,3,6-三氯吡啶還原法的原料不易得,反應條件對設備要求較高,且選擇性不高;2-氯吡啶合成法的目標產物分離困難,收率不高,不具備工業化生產價值;3-氯吡啶合成法所用的原料、試劑成本過高,收率低,沒有優勢;以煙酰胺為原料的合成方法,原料通過霍夫曼降解、氯化,重氮化以及桑德邁爾反應得到2,3-二氯吡啶,此合成路線步驟較多、過程復雜、不易控制且收率較低,產品質量不高。
3、2,3,5-三氯吡啶與堿金屬氫氧化物反應制備3,5-二氯-2-吡啶酚,是合成殺蟲螨以及除草劑噁草醚等系列農藥的重要原料。2,3,5-三氯吡啶也可以進一步進行氟化反應合成2,3-二氟-5-氯吡啶,后者是合成除草劑炔草酯的基礎原料。目前,關于2,3,5-三氯吡啶的合成方法報道不是很多,主要有以下幾種:(1)us4127575和gb1215387a分別公開了以三氯肼基吡啶和3,5-二氯-2-吡啶酮為原料合成2,3,5-三氯吡啶,這兩種方法反應步驟少,收率也比較理想,但是都存在原料昂貴且不易得等缺點;(2)專利201410669523.5公開了2-氯吡啶在堿催化下與水或者醇類反應生成2-烷氧基吡啶,2-烷氧基吡啶在堿基情況下和氯化劑反應生成3,5-二氯-2-烷氧基吡啶,最后氯化得到2,3,5-三氯吡啶,該合成方法步驟繁瑣,產生工業“三廢”比較多,不適合工業化生產;(3)美國專利us4245098公開了一種催化合環法合成2,3,5-三氯吡啶的技術,主要包括以三氯乙醛和丙烯腈為原料,在催化劑的作用下,制備2,3,5-三氯吡啶。該合成工藝操作較為煩瑣,反應條件也較為苛刻,不易控制,且要應用到大量的金屬鹽,會污染環境,同時產品收率也不理想;(4)cn105887128a公開的2,3,5-三氯吡啶的制備方法,是以酸性溶液為反應介質,以五氯吡啶為原料,通過電解反應生成2,3,4,5-四氯吡啶和2,3,5-三氯吡啶,然后進行分離純化;cn108358835a公開的2,3,5-三氯吡啶的制備方法,是以水為溶劑,以碳酸鈉為縛酸劑,以丁醇為催化助劑,將2,3,4,5-四氯吡啶與水合肼進行肼基取代反應,生成三氯吡啶肼基,在堿性物質、相轉移催化劑作用下,經雙氧水直接氧化,得到2,3,5-三氯吡啶。
4、關于此方面的專利均只涉及到一種產品的制備,且過程復雜,效果不佳,因此需要開發一種工藝簡單、綠色環保、成本低廉且產品收率高,純度高的合成工藝。
技術實現思路
1、發明要解決的問題
2、本發明旨在提供一種雙金屬催化劑及其制備方法和應用,所述雙金屬催化劑為含有銀、鎳、鋅、銅、鉻、錳、鈷、鐵或鋯的鈀碳催化劑;該催化劑能夠提高2,3,5,6-四氯吡啶選擇性加氫脫氯制備2,3-二氯吡啶和2,3,5-三氯吡啶的原料轉化率和產物選擇性,同時該催化劑具有優異的穩定性,在多次循環套用后仍能保持較高的活性;該催化劑的制備方法以及利用該催化劑制備2,3-二氯吡啶和2,3,5-三氯吡啶的方法工藝簡單,成本較低,后處理工藝簡單,成本較低,適合工業化生產。
3、用于解決問題的方案
4、為了解決上述問題,本發明提供了一種雙金屬催化劑的制備方法,其包含以下步驟:
5、(1)以糖的水溶液為原料進行水熱碳化反應,冷卻,過濾,干燥,得到納米活性炭;
6、(2)將所述預處理活性炭浸漬于氯化鈀的醇或/和水溶液中,浸漬結束后,干燥,得到催化劑1;
7、(3)將所述催化劑1浸漬于助劑的醇或/和水溶液中,浸漬結束后,煅燒,干燥,即得;
8、其中,所述助劑為硝酸銀、硝酸鎳、氯化鋅、氯化銅、氯化鉻、氯化錳、氯化鈷、氯化鐵或氯化鋯。
9、優選地,步驟(1)中助劑為硝酸鎳。
10、優選地,步驟(1)中所述納米活性炭經sem表征呈球型。普通活性炭經sem表征呈無定形。
11、優選地,步驟(1)中所述納米活性炭的比表面積為500-3000m2/g。在具體實施例中,納米活性炭的比表面積可以為600m2/g、700m2/g、800m2/g、900m2/g、1000m2/g、1100m2/g、1200m2/g、1300m2/g、1400m2/g、1500m2/g、1600m2/g、1700m2/g、1800m2/g、1900m2/g、2000m2/g、2200m2/g、2400m2/g、2600m2/g、2800m2/g或3000m2/g等。
12、優選地,步驟(1)中所述納米活性炭的平均孔徑為1.0-10nm。在具體實施例中,納米活性炭的平均孔徑可以為1.5nm、2.0nm、2.5nm、3.0nm、3.5nm、4.0nm、4.5nm、5.0nm、5.5nm、6.0nm、6.5nm、7.0nm、7.5nm、8.0nm、8.5nm、9.0nm、9.5nm或10nm等。
13、優選地,步驟(1)中所述納米活性炭的孔容為0.5-3.0cm3/g。在具體實施例中,納米活性炭的孔容可以為0.75cm3/g、1.0cm3/g、1.25cm3/g、1.5cm3/g、1.75cm3/g、2.0cm3/g、2.25cm3/g、2.50cm3/g、2.75cm3/g或3.0cm3/g等
14、優選地,步驟(1)中所述納米活性炭的質量比容量為100-450f/g。在具體實施例中,納米活性炭的質量比容量可以為150f/g、200f/g、250f/g、300f/g、350f/g、400f/g或450f/g等。
15、優選地,步驟(1)中所述糖為葡萄糖、果糖、蔗糖或木糖中的一種或一種以上。
16、優選地,步驟(1)中所述糖的水溶液濃度為0.2-0.6mol/l。在具體實施例中,所述糖的水溶液濃度為0.25mol/l、0.3mol/l、0.35mol/l、0.4mol/l、0.45mol/l、0.5mol/l、0.55mol/l或0.6mol/l等。
17、優選地,步驟(1)中所述水熱碳化反應的溫度為150℃-250℃,例如160℃、170℃、180℃、190℃、200℃、210℃、220℃、230℃、240℃或250℃等。
18、優選地,步驟(1)中所述水熱碳化反應的時間為8h-20h,例如9h、10h、11h、12h、13h、14h、15h、16h、17h、18h、19h或20h等。
19、優選地,步驟(1)中所述干燥的溫度為40-80℃,例如45℃、50℃、55℃、60℃、65℃、70℃、75℃或80℃等。
20、優選地,步驟(1)中所述干燥的時間為2-12h,例如3h、4h、5h、6h、7h、8h、9h、10h、11h或12h等。
21、優選地,步驟(2)中所述氯化鈀中的鈀與所述納米活性炭的質量比為2%至10%比1,例如3%:1、4%:1、5%:1、6%:1、7%:1、8%:1、9%:1或10%:1等。
22、優選地,步驟(2)中所述醇為乙醇、甲醇、異丙醇或叔丁醇;
23、優選地,步驟(2)中所述醇水溶液的濃度為60wt%-80wt%,例如60wt%、65wt%、70wt%、75wt%或80wt%等。
24、優選地,步驟(2)中所述浸漬的時間為6-24h,例如6h、8h、10h、12h、14h、15h、16h、18h、20h、22h或24h等。
25、優選地,步驟(2)中所述干燥的溫度為30-50℃,例如30℃、35℃、40℃、45℃或50℃等。
26、優選地,步驟(2)中所述干燥的時間為6-12h,例如6h、7h、8h、9h、10h、11h或12h等。
27、優選地,步驟(3)中所述助劑中的金屬與步驟(2)中所述氯化鈀中的鈀的摩爾比為0.01-0.1:1,優選0.05-0.1:1,更優選0.07:1;例如0.01:1、0.02:1、0.03:1、0.04:1、0.05:1、0.06:1、0.07:1、0.08:1、0.09:1或0.1:1等。
28、優選地,步驟(3)中所述浸漬的時間為6-24h,例如6h、8h、10h、12h、14h、15h、16h、18h、20h、22h或24h等。
29、優選地,步驟(3)中所述煅燒溫度為300-400℃,例如300℃、320℃、340℃、350℃、360℃、380℃或400℃等。
30、優選地,步驟(3)中所述煅燒時間為2-4h,例如2h、2.5h、3h、3.5h或4h等。
31、優選地,步驟(3)中所述干燥的溫度為30-50℃,例如30℃、35℃、40℃、45℃或50℃等。
32、優選地,步驟(3)中所述干燥的時間為6-12h,例如6h、7h、8h、9h、10h、11h或12h等。
33、另一方面,本發明提供了一種雙金屬催化劑,其由上述制備方法得到。
34、再一方面,本發明提供了上述雙金屬催化劑在脫氯加氫反應中的應用。
35、優選地,所述脫氯加氫反應的原料為被氯原子取代的芳環或雜芳環化合物。
36、更優選地,所述脫氯加氫反應為由2,3,5,6-四氯吡啶制備2,3,5-三氯吡啶和2,3-二氯吡啶的反應。
37、再一方面,本發明提供了一種聯產2,3,5-三氯吡啶和2,3-二氯吡啶的制備方法,其包括以下步驟:在有機溶劑和縛酸劑的存在下,以2,3,5,6-四氯吡啶為原料,以氫氣為氫源,在前述雙金屬催化劑的催化下反應,得到2,3,5-三氯吡啶和2,3-二氯吡啶。
38、優選地,所述縛酸劑選自無機堿,有機堿,磷酸鹽或羧酸鹽中的一種或多種。
39、優選地,所述無機堿為堿金屬氫氧化物、堿金屬氧化物、堿金屬碳酸鹽、堿金屬碳酸氫鹽、堿土金屬碳酸鹽、堿土金屬碳酸氫鹽、堿土金屬氧化物、堿土金屬氫氧化物、碳酸銨、碳酸氫銨或酸式鹽中的一種或多種,優選堿金屬碳酸鹽、堿土金屬氧化物。
40、進一步地,所述無機堿選自碳酸鈉、碳酸鉀、碳酸氫鈉、碳酸氫鉀、碳酸氫銨、氧化鎂、氫氧化鎂、氧化鈣、氫氧化鈣中的一種或多種,優選碳酸鈉或氧化鎂,更優選氧化鎂。
41、所述有機堿選自吡啶、烷基吡啶或有機胺中的一種或多種。
42、進一步地,所述烷基吡啶選自2-甲基吡啶、3-甲基吡啶或4-甲基吡啶中的一種或多種。
43、進一步地,所述有機胺選自脂肪胺、脂環胺或芳香胺中的一種或多種。
44、更進一步地,所述有機胺選自甲胺、乙胺、丙胺、二甲胺、乙二胺、二乙胺或三乙胺中的一種或多種。
45、優選地,所述磷酸鹽選自磷酸二氫鹽、磷酸氫鹽或正磷酸鹽中的一種或多種。
46、進一步地,所述磷酸鹽選自磷酸二氫鈉、磷酸二氫鉀、磷酸鉀、磷酸二氫銨、磷酸氫二銨、連二磷酸銨、磷酸三銨中的一種或多種。
47、優選地,所述羧酸鹽選自甲酸鹽、乙酸鹽、丙三酸鹽和丁四酸鹽中的一種或多種。
48、進一步地,所述羧酸鹽選自甲酸銨、甲酸鈉、甲酸鉀、乙酸銨、乙酸鈉中的一種或多種。
49、優選地,所述縛酸劑與所述2,3,5,6-四氯吡啶的摩爾比為0.2:1-3:1。在具體實施例中,所述縛酸劑與所述2,3,5,6-四氯吡啶的摩爾比可以為0.4:1、0.6:1、0.8:1、1.0:1、1.2:1、1.4:1、1.6:1、1.8:1、2.0:1、2.2:1、2.4:1、2.6:1、2.8:1或3.0:1等。
50、優選地,所述2,3,5,6-四氯吡啶與所述雙金屬催化劑的質量比為1:0.001-1:0.01。在具體實施例中,所述2,3,5,6-四氯吡啶與所述雙金屬催化劑的質量比為1:0.002、1:0.003、1:0.004、1:0.005、1:0.006、1:0.007、1:0.008、1:0.009或1:0.01等。
51、優選地,所述有機溶劑選自含苯環化合物類,呋喃類化合物,酯類溶劑、醇類溶劑,腈類溶劑或烷烴類溶劑中的一種或一種以上。
52、進一步地,所述有機溶劑選自:c6-c12的含苯環化合物,例如甲苯、二甲苯、三甲苯、對二甲苯等;c4-c8的呋喃類化合物,例如四氫呋喃;烷羧酸烷醇酯,尤其是c1-c8烷羧酸與c1-c8烷醇的酯,例如乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酸丁酯等;c1~c8醇,例如甲醇、乙醇、丙醇、異丙醇等;c2~c4腈,例如乙腈、丙腈;c6~c10烷烴,例如正己烷、環己烷等。
53、優選地,所述有機溶劑與所述2,3,5,6-四氯吡啶的質量比為1:1-10:1。在具體實施例中,有機溶劑與2,3,5,6-四氯吡啶的質量比可以在1:1至10:1的范圍內。在某些具體實施例中,有機溶劑與2,3,5,6-四氯吡啶的質量比可以為1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1等。在一個具體實施例中,有機溶劑與2,3,5,6-四氯吡啶的質量比4比1。
54、優選地,所述氫氣的壓力為0.1-3mpa。在具體實施例中,氫氣的壓力可以為0.2mpa、0.3mpa、0.4mpa、0.5mpa、0.6mpa、0.7mpa、0.8mpa、0.9mpa、1.0mpa、1.2mpa、1.4mpa、1.5mpa、1.6mpa、1.8mpa、2.0mpa、2.2mpa、2.4mpa、2.5mpa、2.6mpa、2.8mpa或3.0mpa等。
55、優選地,所述反應的溫度為20-100℃,例如在25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃、90℃或100℃等的溫度下進行。
56、優選地,所述反應的時間沒有特別的限制,只要加氫催化反應基本完成即可。“基本完成”是式(i)化合物的轉化率大于等于80%。優選地,本發明的方法的反應時間可以在2小時至14小時范圍內,例如3小時、4小時、5小時、6小時、7小時、8小時、9小時、10小時、12小時或14小時等。
57、優選地,所述反應還包括后處理步驟,所述后處理步驟包括過濾、蒸餾和精餾等步驟。
58、進一步地,所述蒸餾在常壓下進行,蒸餾溫度為40℃至100℃。
59、進一步地,所述精餾在真空度≤-0.07mpa下進行,精餾溫度為100℃至150℃,回流比在5至20比1。
60、具體到實施例中,先向高壓釜中加入2,3,5,6-四氯吡啶、催化劑,再加入有機溶劑和縛酸劑;通入氮氣進行置換3次,最后通入氫氣進行置換;置換完成后,保持高壓釜內氫氣壓力在0.1-3.0mpa之間,同時控制溫度保持在20-100℃內,開啟攪拌,反應時長在2-14h;待反應結束,自然降至室溫,排出釜內殘留氣體,并通入氮氣吹掃;對反應液進行過濾,分離催化劑,濾液經常壓蒸餾分離溶劑,再通過精餾分離得到2,3-二氯吡啶和2,3,5-三氯吡啶。
61、本發明與現有技術相比,具備以下幾點優勢:
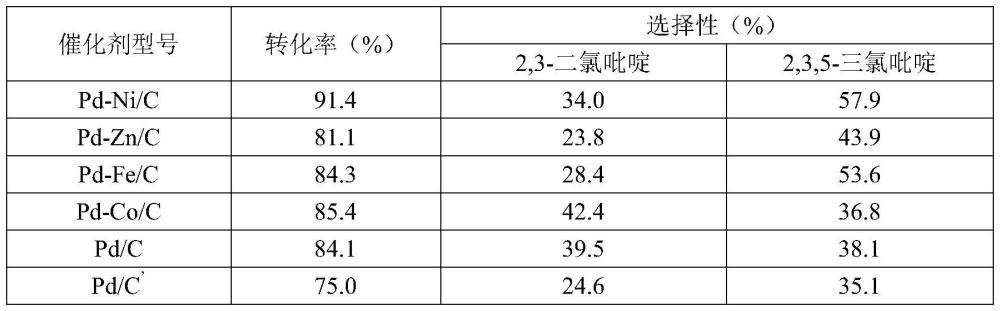
62、1、本發明通過采用2,3,4,5-四氯吡啶制備2,3-二氯吡啶和2,3,5-三氯吡啶,使用氫氣作為氫源,在催化劑及縛酸劑的作用下,選擇性脫氯獲得2,3-二氯吡啶和2,3,5-三氯吡啶,原料能夠完全轉化,且兩種產物的收率高達96%以上;
63、2、2,3,5,6-四氯吡啶作為吡啶氯化過程中的副產物,利用價值低,本發明以2,3,5,6-四氯吡啶為原料,制備更高價值的2,3-二氯吡啶和2,3,5-三氯吡啶,且工藝簡單,性能好,具有極大工業價值;
64、3、本發明中的催化劑采用pd作為活性組分,通過金屬間相互協同作用,進一步提高的反應性能,同時能夠減少催化劑使用量以及延長了催化劑使用壽命,降低了生產成本;
65、4、本發明反應體系中堿的加入,有效解決了反應過程中生成的hcl對反應釜的腐蝕,以及對催化劑破壞,且反應工藝條件較為溫和,反應徹底,無過多三廢生成,方法經濟環保。
- 還沒有人留言評論。精彩留言會獲得點贊!