一種調變單斜相/六方相異相結三氧化鎢光催化劑中六方相含量的方法與流程
本發明屬于材料領域,尤其涉及一種調控異相結型光催化劑的光催化性能的制備方法,具體是一種調變單斜相/六方相三氧化鎢異相結中六方相(或單斜相)含量的方法,從而調控單斜相/六方相三氧化鎢異相結催化劑的光催化性能。
背景技術:
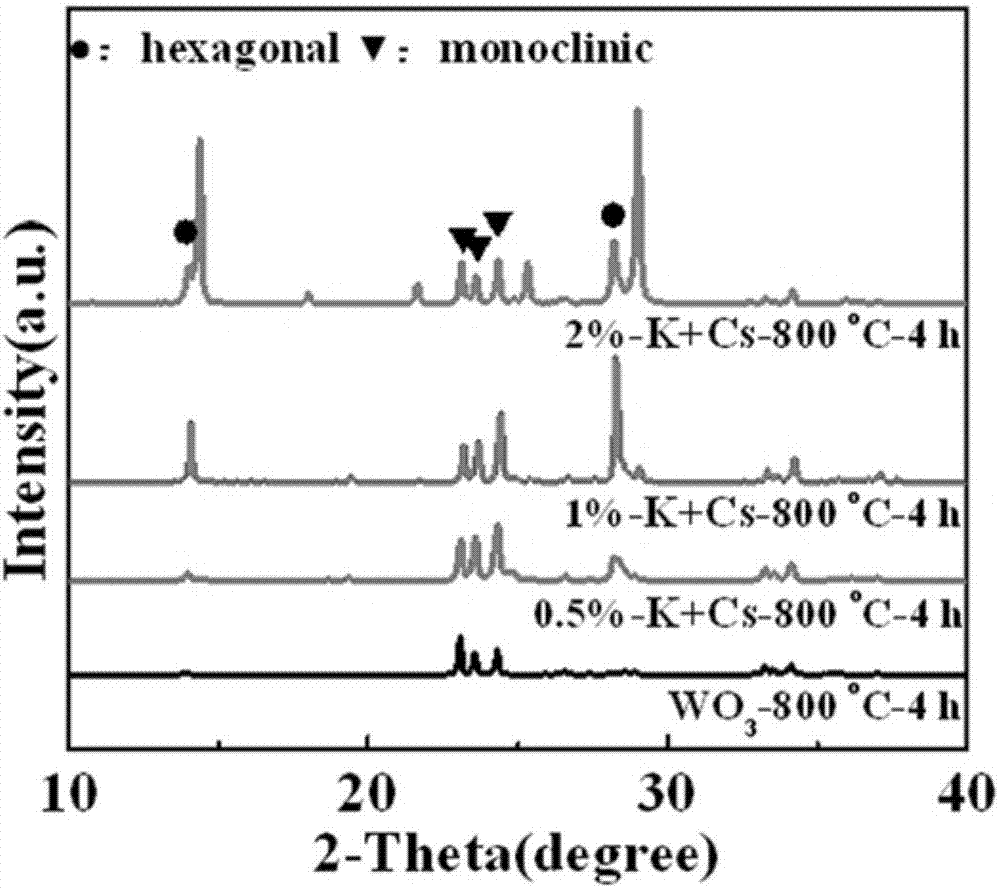
:近年來,作為一種n型半導體,三氧化鎢(wo3)因其在光催化降解有機污染物和光催化分解水方面顯示出優異性能而備受關注。wo3禁帶寬度較窄(2.4-2.8ev),有利于可見光的吸收,與光催化領域中廣泛使用的tio2相比,對于太陽能中可見光的利用率顯著提高。然而,較高的光生電子-空穴復合效率阻礙wo3光催化性能的進一步提高。通常采取增大比表面積、減小粒徑、元素摻雜、與其他金屬氧化物耦合形成“異質結”等方式改善wo3光催化性能。研究表明,異相結(不同晶相之間形成的界面)的形成也能夠有效地促進光生電子-空穴分離,從而提高光催化性能。liu等(liu,z.;zhang,x.;jin,m.;tryk,d.a.anatasetio2nanoparticlesonrutiletio2nanorods:aheterogeneousnanostructurevialayer-by-layerassembly.langmuir.2007,23,10916-10919.),研究了調變銳鈦礦/金紅石二氧化鈦“異相結”中兩相含量對其光催化性能的影響,結果表明合適的相比例,有效地促進光生電子-空穴分離,從而導致光催化活性提高1.2倍,這為我們進一步改善wo3光催化性能提供思路。wo3具有單斜、三斜、正交、六方、立方等晶相,其中單斜相與六方相是廣泛應用在光催化領域的兩種晶相。我們課題組的研究(lu,y.;liu,g.;zhang,j.;feng,z.;li,c.;li,z.fabricationofamonoclinic/hexagonaljunctioninwo3anditsenhancedphotocatalyticdegradationofrhodamineb.chinesej.catal.2016,37,349-358)結果表明,單斜相和六方相wo3之間“異相結”的形成能夠很好地促進wo3半導體表面光生電子-空穴的分離,與單一晶相(純單斜相)wo3相比,形成“異相結”后光催化性能提高到原來的2倍。然而,僅僅通過簡單的改變焙燒條件很難對“異相結”中兩相含量進行調變,六方相含量最高只有21.36%。熱力學上,單斜相為穩相,而六方相屬于亞穩相。優先制得六方相wo3,在六方相逐漸轉變為單斜相的過程中構建單斜相/六方相異相結更容易實現對異相結中兩相含量調變的目的。然而研究表明,六方相wo3需要外加離子或分子來穩定其六方相孔道結構,并且一旦這些穩定離子或分子流失,那么骨架就會坍塌,立即轉變為單斜相。因此,如何穩定六方相,并在焙燒時控制六方相向單斜相轉化是調變單斜相/六方相異相結中六方相含量的關鍵步驟。技術實現要素:本發明針對上述的問題,提供一種調變單斜相/六方相異相結wo3光催化劑中六方相(單斜相)含量的簡單方法,從而實現調控單斜相/六方相wo3異相結催化劑的光催化性能。本發明操作簡單,不僅能夠實現六方相含量在0-71.1%之間連續調變,并且制備的單斜相/六方相最優比例(六方相含量為71.1%)異相結wo3光催化劑的光催化性能是單一晶相(純單斜相)wo3光催化劑的2.5倍。為了實現上述目的,本發明提供的調變單斜相/六方相異相結wo3光催化劑中六方相(單斜相)含量的方法,具體包括以下步驟。步驟1、將鎢酸鹽與去離子水混合,配制成鎢酸鹽含量為2-10wt%的懸濁液;向鎢酸鹽懸濁液中加入一定量的晶型穩定劑,再加入一定量絡合劑;待上述溶液變成澄清透明溶液之后,再加入10ml乙二醇。步驟2、將上述混合溶液在70-90℃下濃縮,得到濕凝膠;將濕凝膠經60℃-120℃干燥后得到干凝膠;將干凝膠放入馬弗爐,在350-450℃焙燒1.5-4h,研磨成干凝膠粉末。步驟3、對步驟2得到的干凝膠粉末,在700-900℃條件下再次進行焙燒2-54h,得到單斜相/六方相異相結wo3催化劑。所述鎢酸鹽為鎢酸銨、偏鎢酸銨或鎢酸鈉。優選的,所述晶型穩定劑為k2so4和cs2so4的混合晶型穩定劑,其中k2so4與cs2so4的摩爾比為1:1。所述晶型穩定劑還可以為k2so4、cs2so4、k2c2o4、na2so4或na2c2o4中的一種或者組合。所述晶型穩定劑中堿金屬離子的質量與鎢酸鹽中鎢的質量比為0.5-2:100,優選1:100。所述絡合劑為葡萄糖酸鈉、酒石酸、草酸或檸檬酸,優選葡萄糖酸鈉。所述鎢酸鹽中鎢的物質的量與絡合劑的物質的量的比是2:1。優選的,步驟2中得到的混合溶液在90℃下濃縮,得到濕凝膠。優選的,步驟2中焙燒溫度為350℃,時間為1.5h。優選的,步驟3中焙燒溫度優選800℃,焙燒時間為2-54h;當焙燒溫度為700℃時,焙燒時間為4-20h;當焙燒溫度為900℃時,焙燒時間為4-25h。本發明的有益效果。本發明在添加晶型穩定劑的條件下,僅僅采用改變焙燒溫度和焙燒時間的方法,可以實現單斜相/六方相異相結中六方相含量的調變目的;通過對絡合劑種類、晶型穩定劑種類、晶型穩定劑添加量、焙燒溫度和焙燒時間的考察發現,當絡合劑為葡萄糖酸鈉、晶型穩定劑為k2so4與cs2so4的混合晶型穩定劑(摩爾比1:1)、添加量為1wt%(晶型穩定劑中堿金屬離子與鎢酸鹽中鎢的質量比為1:100)、焙燒溫度為700-900℃、焙燒時間為2-54h時六方相含量在0-71.1%區間內可連續調變;并且研究發現當采用其他晶型穩定劑如k2so4、cs2so4、k2c2o4、na2so4或na2c2o4,在最優條件下(晶型穩定劑堿金屬加入量為1wt%,絡合劑為葡萄糖酸鈉,焙燒溫度為800℃焙燒時間為4h)考察,發現不能進行調變。進一步研究了本發明調變得到的具有不同晶相組成的異相結wo3催化劑樣品的光催化性能,當兩相最優比(六方相為71.1%)時,樣品的光催化性能為純單斜相樣品的2.5倍。目前,對于單斜相/六方相異相結wo3催化劑的研究很有限,更沒有報道調變異相結中兩相含量的研究,而本發明的研究剛好填補了這方面的空白。本發明采用溶膠凝膠法考察了是否加入晶型穩定劑、加入晶型穩定劑的種類、晶型穩定劑的加入量、絡合劑的種類、焙燒溫度以及焙燒時間,對單斜相/六方相異相結wo3催化劑中六方相含量的影響。結果表明當加入k2so4與cs2so4混合晶型穩定劑(摩爾比為1:1)且加入量為1wt%(晶型穩定劑中堿金屬離子與鎢酸鹽中鎢的質量比為1:100),絡合劑為葡萄糖酸鈉(鎢酸鹽中鎢與絡合劑的摩爾比為2:1)時,通過在700-900℃焙燒2-54h的方式可以將單斜相/六方相異相結wo3催化劑中六方相含量在0-71.1%區間內進行調變。而未加晶型穩定劑的wo3催化劑的六方相含量最高只有9.3%,不能進行調變;加入晶型穩定劑分別為k2so4、cs2so4、k2c2o4、na2so4、na2c2o4制備得到的wo3催化劑的六方相含量最高只有48.2%、53.5%、26.5%、31.4%、35.8%,不能進行調變。加入混合晶型穩定劑k2so4與cs2so4很好地穩定了六方相,控制了六方相向單斜相轉變,使得簡單通過改變焙燒溫度、焙燒時間就能實現單斜相/六方相異相結wo3催化劑中六方相含量的調變,從而實現了對單斜相/六方相異相結wo3催化劑光催化性能的調控,在最優的比例(六方相為71.1%)下得到的單斜相/六方相wo3異相結催化劑樣品光催化降解效率是純相(純單斜相)wo3催化劑樣品的2.5倍。這是因為k+、na+離子半徑過小可以在六方相的三方孔道和六方孔道中同時存在,在焙燒過程中離子發生遷移,六方相框架發生改變,對六方相的穩定性不佳;而cs+的離子半徑又過大不易轉化為單斜相,改變焙燒條件調變六方相含量困難;而當k+與cs+相互配合時既能夠很好的穩定六方相又能夠在焙燒時逐步轉化為單斜相,達到調變含量的目的。此外我們還考察了絡合劑種類對于單斜相/六方相異相結wo3催化劑中六方相含量的影響,結果發現,當絡合劑為葡萄糖酸鈉時,可以通過改變焙燒溫度和焙燒時間使六方相在0-71.1%區間內調變;而絡合劑為酒石酸、草酸、檸檬酸時,相同焙燒條件下六方相含量最高分別為3.4%、7.1%、9.2%且不能調變。這是因為在此種體系中葡萄糖酸鈉的絡合作用最優,能夠穩定六方相,控制六方相向單斜相的轉化,使得通過改變焙燒溫度和時間就能達到調變六方相含量的作用。同時,本發明采用溶膠凝膠法進行制備,改變焙燒溫度和焙燒時間進行含量調變,所使用的原料價格低廉,制備過程操作簡便,無需高壓條件和專有設備,工藝簡單,制備成本低。附圖說明圖1(a)為加入混合晶型穩定劑k2so4與cs2so4制得的1%-k+cs干凝膠粉末在700-900℃焙燒4h,得到樣品的x射線衍射(xrd)圖;(b)為不加晶型穩定劑制得的p-wo3干凝膠粉末在700-900℃焙燒4h,得到樣品的x射線衍射(xrd)圖。圖2為加入不同量混合晶型穩定劑k2so4與cs2so4(摩爾比為1:1)制得m%-k+cs干凝膠粉末(混合晶型穩定劑k2so4與cs2so4中堿金屬離子與鎢酸鹽中鎢的質量比為m:100)在800℃下焙燒4h,得到樣品的x射線衍射(xrd)圖。圖3為1%-k+cs干凝膠粉末在800℃焙燒不同時間,制備得到的具有不同六方相含量的單斜相/六方相異相結樣品的x射線衍射(xrd)圖。圖4為1%-k+cs干凝膠粉末在800℃焙燒不同時間,制備得到的具有不同六方相含量的單斜相/六方相異相結樣品對羅丹明b分解轉化率隨時間變化圖。圖5為對比例1中分別以k2so4、cs2so4為晶型穩定劑制備得到的1%-k2so4、1%-cs2so4干凝膠粉末在800℃焙燒4h,得到樣品的x射線衍射(xrd)圖。圖6為對比例2中分別以k2c2o4、na2so4、na2c2o4為晶型穩定劑制備得到的1%-k2c2o4、1%-na2so4、1%-na2c2o4干凝膠粉末在800℃焙燒4h,得到樣品的x射線衍射(xrd)圖。圖7為對比例3中分別以酒石酸、草酸、檸檬酸為絡合劑制備得到的1%-k+cs-酒石酸、1%-k+cs-草酸、1%-k+cs-檸檬酸干凝膠粉末在800℃焙燒4h,得到樣品的x射線衍射(xrd)圖。具體實施方式下面結合具體實施例對本發明做詳細的說明。實施例1。以k2so4與cs2so4為混合晶型穩定劑改變焙燒溫度調變單斜相/六方相異相結wo3催化劑中六方相含量的制備方法,包括如下步驟。(1)將10g鎢酸銨與190ml水混合形成懸濁液;向鎢酸銨懸濁液中加入0.0366gk2so4和0.0760gcs2so4(k2so4與cs2so4摩爾比為1:1)(k2so4與cs2so4中k+與cs+的質量和與鎢酸銨中鎢的質量比為1:100),再加入4.3017g葡萄糖酸鈉(鎢酸銨中鎢的物質的量與葡萄糖酸鈉的物質的量的比是2:1);待上述溶液變成澄清透明溶液之后再加入10ml乙二醇。(2)將上述混合溶液在90℃下濃縮,得到濕凝膠;將濕凝膠經60℃干燥后得到干凝膠;將干凝膠放入馬弗爐,在350℃焙燒1.5h,研磨成粉末標記為1%-k+cs干凝膠粉末。(3)對步驟(2)得到的1%-k+cs干凝膠粉末樣品,在700-900℃再次進行焙燒4h,得到單斜相/六方相異相結wo3催化劑,分別標記為1%-k+cs-700℃-4h、1%-k+cs-800℃-4h、1%-k+cs-900℃-4h。(4)為了形成對比研究,參照步驟(1)-(3)制備不加晶型穩定劑k2so4與cs2so4的wo3光催化劑,不同之處在于不加入晶型穩定劑且步驟(2)得到的干凝膠粉末標記為p-wo3干凝膠粉末,步驟(3)得到的樣品分別標記為wo3-700℃-4h、wo3-800℃-4h、wo3-900℃-4h。圖1中(a)是加入晶型穩定劑k2so4+cs2so4制得的1%-k+cs干凝膠粉末在700-900℃焙燒4h得到樣品的x射線衍射(xrd)圖;圖1(b)為不加晶型穩定劑制得的p-wo3干凝膠粉末在700-900℃焙燒4h得到樣品的x射線衍射(xrd)圖。在2θ=23.1°,23.6°,24.4°處觀察到屬于單斜相wo3的衍射峰,在2θ=14.0°,28.2°處觀察到屬于六方相wo3的衍射峰,且兩個晶相的衍射峰強度不同,說明樣品中既含有單斜相也含有六方相,且兩相所占的比例不同。利用下列公式計算wo3異相結樣品中單斜相與六方相的含量。wm=1/[1+0.679(ih/im)](1)。wh=1-wm(2)。其中,wm和wh分別表示單斜相和六方相的含量(%),im和ih分別表示單斜相(002)和六方相(200)衍射峰的強度。通過公式(1)、(2)計算得到的六方相含量如表1所示。表11%-k+cs干凝膠粉末和p-wo3干凝膠粉末在700-900℃焙燒4h得到樣品的六方相含量表。在不同溫度焙燒條件下加入晶型穩定劑k2so4與cs2so4制得的具有單斜相/六方相wo3異相結結構的催化劑,通過改變焙燒溫度,在700-900℃焙燒4h使得六方相含量在0-71.1%之間可調,而未加k2so4與cs2so4制得的催化劑六方相含量在相同的焙燒條件下六方相含量最高僅為9.3%,并且不能調變,說明相同焙燒條件下,混合晶型穩定劑k2so4與cs2so4的加入穩定了六方相,控制了六方相向單斜相的轉化。實施例2。以k2so4與cs2so4為混合晶型穩定劑,改變混合晶型穩定劑k2so4與cs2so4(摩爾比為1:1)加入量m:100=0.5:100-2:100(混合晶型穩定劑中堿金屬離子與鎢酸鹽中鎢的質量比為m:100)調變單斜相/六方相異相結wo3催化劑中六方相含量的制備方法包括如下步驟。(1)將10g鎢酸銨與190ml水混合形成懸濁液。向鎢酸銨懸濁液中加入0.0183gk2so4和0.0380gcs2so4(0.5:100)、0.0366gk2so4和0.0760gcs2so4(1:100)、0.0732gk2so4和0.1520gcs2so4(2:100)(摩爾比為1:1)(k2so4+cs2so4中k+與cs+的質量和與鎢酸銨中鎢的質量比為0.5:100-2:100),再加入4.3017g葡萄糖酸鈉(鎢酸銨中鎢的物質的量與葡萄糖酸鈉的物質的量的比是2:1);待上述溶液變成澄清透明溶液之后再加入10ml乙二醇。(2)將上述混合溶液在90℃下濃縮,得到濕凝膠;將濕凝膠經60℃干燥后得到干凝膠;將干凝膠放入馬弗爐,在350℃焙燒1.5h,研磨成粉末標記為m%-k+cs干凝膠粉末(混合晶型穩定劑中堿金屬離子與鎢酸鹽中鎢的質量比為m:100,其中k2so4與cs2so4為摩爾比1:1混合)。(3)對步驟(2)得到的m%-k+cs干凝膠粉末樣品,在800℃再次進行焙燒4h,得到單斜相/六方相異相結wo3催化劑,分別標記為0.5%-k+cs-800℃-4h、1%-k+cs-800℃-4h、2%-k+cs-800℃-4h(百分數指的是混合晶型穩定劑中堿金屬離子與鎢酸鹽中鎢的質量百分比)。圖2是加入不同量k2so4與cs2so4混合晶型穩定劑制得m%-k+cs干凝膠粉末在800℃下焙燒4h得到樣品的x射線衍射(xrd)圖,在2θ=23.1°,23.6°,24.4°處觀察到屬于單斜相wo3的衍射峰,在2θ=14.0°,28.2°處觀察到屬于六方相wo3的衍射峰,且兩個晶相的衍射峰強度不同,說明樣品中既含有單斜相也含有六方相,且兩相所占的比例不同。通過實施例1中公式(1)、(2)計算得到的六方相含量如表2所示。表2m%-k+cs干凝膠粉末在800℃下焙燒4h得到樣品的六方相含量表。樣品h含量樣品h含量wo3-800℃-4h3.9%1%-k+cs-800℃-4h71.1%0.5%-k+cs-800℃-4h29%2%-k+cs-800℃-4h51.6%當k2so4與cs2so4的加入量由0.5wt%增加到2wt%時,得到的單斜相/六方相異相結wo3催化劑中六方相含量在29-71.1%之間可調變,當混合晶型穩定劑的加入量超過1wt%時,六方相含量下降,并且在六方相2θ=14.0°處衍射峰右側出現強度很高的雜質峰;因此,穩定六方相最適宜的k2so4與cs2so4加入量是1wt%。實施例3。以k2so4與cs2so4為混合晶型穩定劑800℃下,改變焙燒時間調變單斜相/六方相異相結wo3催化劑中六方相含量的制備方法,包括如下步驟。步驟同實施例1,不同之處在于步驟(3)為將步驟(2)中制得的1%-k+cs干凝膠粉末在800℃進行焙燒2h、4h、24h、54h,分別標記為1%-k+cs-800℃-2h、1%-k+cs-800℃-4h、1%-k+cs-800℃-24h、1%-k+cs-800℃-54h。圖3是1%-k+cs干凝膠粉末在800℃焙燒不同時間制備得到的具有不同六方相含量的單斜相/六方相異相結樣品的x射線衍射(xrd)圖,在2θ=23.1°,23.6°,24.4°處觀察到屬于單斜相wo3的衍射峰,在2θ=14.0°,28.2°處觀察到屬于六方相wo3的衍射峰,且兩個晶相的衍射峰強度不同,說明樣品中既含有單斜相也含有六方相,且兩相所占的比例不同。通過實施例1中公式(1)、(2)計算得到的六方相含量如表3所示。表31%-k+cs干凝膠粉末在800℃焙燒2-54h得到樣品的六方相含量表。樣品h含量樣品h含量wo3-800℃-4h3.9%1%-k+cs-800℃-24h59.5%1%-k+cs-800℃-2h28.1%1%-k+cs-800℃-54h01%-k+cs-800℃-4h71.1%--800℃下通過改變焙燒時間,隨著焙燒時間從2h增加到54h單斜相/六方相異相結wo3催化劑中六方相含量分別為28.1%、71.1%、59.5%、0,呈現先增加后減少的趨勢,并且在0-71.1%之間能夠進行調控。此外,還對700℃和900℃進行焙燒時間的考察,發現700℃焙燒4-20h能夠實現六方相含量0-28.2%的調變,900℃焙燒4-25h能夠實現0-30.9%的調變。因此通過改變焙燒溫度700-900℃和焙燒時間2-54h,能夠實現六方相含量在0-71.1%區間的調變。二、以k2so4與cs2so4為晶型穩定劑,800℃焙燒不同時間制備得到的具有不同六方相含量的單斜相/六方相異相結樣品的光催化活性。利用光催化降解羅丹明b為模型反應,考察了以k2so4與cs2so4為混合晶型穩定劑,800℃焙燒不同時間制備得到的具有不同六方相含量的單斜相/六方相異相結樣品的光催化活性。將125w汞燈光源懸于容積為250ml的燒杯上方。將0.05g催化劑超聲分散在60ml初始濃度為10mg/l羅丹明b溶液中攪拌形成懸浮體系,利用125w汞燈光源進行光降解反應。在黑暗條件將反應溶液攪拌30min以達到吸附平衡,隨后進行光照。光照開始后,每隔一定時間取相同體積的試樣溶液,離心后取上層清夜在羅丹明b的553nm吸收波長處測定其吸光度值,根據標準曲線確定羅丹明b的濃度。分析方法:在羅丹明b的最大吸收波長(553nm)處對離心得到的上層清液中羅丹明b的濃度進行測試,根據朗伯-比爾定律,濃度與吸光度成正比,羅丹明b的光致降解率d可由下式求出。其中,ao為光照前暗吸附達到平衡后羅丹明b的吸光度,a為光照時間為t時羅丹明b的吸光度。圖4是實施例3中1%-k+cs干凝膠粉末在800℃焙燒不同時間制備得到的具有不同六方相含量的單斜相/六方相異相結樣品對羅丹明b分解轉化率隨時間變化圖;從圖中我們可以看出經過180min光照以后,1%-k+cs-800℃-4h異相結樣品的光催化活性最高達到68%,與1%-k+cs-800℃-54h純單斜相樣品相比光催化活性提高2.5倍,并且光催化活性隨著異相結中六方相含量的增加而升高。對比例1。參照實施例1中制備單斜相/六方相異相結wo3光催化劑的方法,不同之處在于步驟(1)中,將混合晶型穩定劑分別改為k2so4、cs2so4,步驟(2)中制得的干凝膠粉末標記為1%-k2so4干凝膠粉末、1%-cs2so4干凝膠粉末,步驟(3)為將1%-k2so4干凝膠粉末、1%-cs2so4干凝膠粉末在800℃焙燒4h,得到單斜相/六方相異相結wo3催化劑,分別標記為1%-k2so4-800℃-4h、1%-cs2so4-800℃-4h。圖5是分別以k2so4、cs2so4為晶型穩定劑制備得到的1%-k2so4、1%-cs2so4干凝膠粉末在800℃焙燒4h得到樣品的x射線衍射(xrd)圖。可以觀察到屬于單斜相wo3的衍射峰和屬于六方相wo3的衍射峰,且兩個晶相的衍射峰強度不同,說明樣品中既含有單斜相也含有六方相,且兩相所占的比例不同,通過實施例1中公式計算得到六方相含量如表4所示。但是晶型穩定劑為k2so4、cs2so4時,相同焙燒條件下單斜相/六方相異相結wo3催化劑中六方相含量最高分別為48.2%、53.5%,并且不能通過改變焙燒條件而進行調控。表41%-k2so4、1%-cs2so4干凝膠粉末在800℃焙燒4h得到樣品的六方相含量表。樣品1%-k+cs-800℃-4h1%-k2so4-800℃-4h1%-cs2so4-800℃-4hh含量71.1%48.2%53.5%對比例2。參照實施例1中制備單斜相/六方相異相結wo3光催化劑的方法,不同之處在于步驟(1)中,將混合晶型穩定劑分別改為k2c2o4、na2so4、na2c2o4,步驟(2)中制得的干凝膠粉末分別標記為1%-k2c2o4、1%-na2so4、1%-na2c2o4干凝膠粉末,步驟(3)為分別將1%-k2c2o4、1%-na2so4、1%-na2c2o4干凝膠粉末在800℃焙燒4h,得到單斜相/六方相異相結wo3催化劑,分別標記為1%-k2c2o4-800℃-4h、1%-na2so4-800℃-4h、1%-na2c2o4-800℃-4h。圖6是分別以k2c2o4、na2so4、na2c2o4為晶型穩定劑制備得到的1%-k2c2o4、1%-na2so4、1%-na2c2o4干凝膠粉末在800℃焙燒4h得到樣品的x射線衍射(xrd)圖。可以觀察到屬于單斜相wo3的衍射峰和屬于六方相wo3的衍射峰,且兩個晶相的衍射峰強度不同,說明樣品中既含有單斜相也含有六方相,且兩相所占的比例不同,通過實施例1中公式計算得到六方相含量如表5所示。但是晶型穩定劑分別為k2c2o4、na2so4、na2c2o4時,相同焙燒條件下單斜相/六方相異相結wo3催化劑中六方相含量最高分別為26.5%、31.4%、35.8%,并且不能通過改變焙燒條件而進行調控。同時當晶型穩定劑中堿金屬離子為na+時出現了屬于na2w4o13的衍射峰。表5為1%-k2c2o4、1%-na2so4、1%-na2c2o4干凝膠粉末在800℃焙燒4h得到樣品的六方相含量表。樣品1%-k2c2o4-800℃-4h1%-na2so4-800℃-4h1%-na2c2o4-800℃-4hh含量26.5%31.4%35.8%對比例3。參照實施例1中制備單斜相/六方相異相結wo3光催化劑的方法,不同之處在于步驟(1)中,將絡合劑葡萄糖酸鈉分別改為酒石酸、草酸、檸檬酸,步驟(2)中制得的干凝膠粉末分別標記為1%-k+cs-酒石酸、1%-k+cs-草酸、1%-k+cs-檸檬酸干凝膠粉末,步驟(3)為分別將1%-k+cs-酒石酸、1%-k+cs-草酸、1%-k+cs-檸檬酸干凝膠粉末在800℃焙燒4h,得到單斜相/六方相異相結wo3催化劑,分別標記為1%-k+cs-酒石酸-800℃-4h、1%-k+cs-草酸-800℃-4h、1%-k+cs-檸檬酸-800℃-4h。圖7是分別以酒石酸、草酸、檸檬酸為絡合劑制備得到的1%-k+cs-酒石酸、1%-k+cs-草酸、1%-k+cs-檸檬酸干凝膠粉末在800℃焙燒4h得到樣品的x射線衍射(xrd)圖。可以觀察到屬于單斜相wo3的衍射峰和屬于六方相wo3的衍射峰,且兩個晶相的衍射峰強度不同,說明樣品中既含有單斜相也含有六方相,且兩相所占的比例不同,通過實施例1中公式計算得到六方相含量如表6所示。但是絡合劑分別改為酒石酸、草酸、檸檬酸時,相同焙燒條件下單斜相/六方相異相結wo3催化劑中六方相含量最高分別為3.4%、7.1%、9.2%,并且不能通過改變焙燒條件而進行調控。表6為1%-k+cs-酒石酸、1%-k+cs-草酸、1%-k+cs-檸檬酸干凝膠粉末在800℃焙燒4h得到樣品的六方相含量表。樣品1%-k+cs-酒石酸1%-k+cs-草酸1%-k+cs-檸檬酸h含量3.4%7.1%9.2%本發明的上述實施例僅為說明本發明所作的舉例,而并非是對本發明的具體實施方式的限定。對于所屬領域的普通技術人員來說,在上述舉例的基礎上還可以做其他不同形式的變化或變動。這里無法對所有的實施方式予以詳細舉例。凡是屬于本發明的技術方案所引申出的顯而易見的變化或變動仍處于本發明的保護范圍之列。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1