一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法與流程
本發明屬于喜樹堿的檢測技術領域,特別涉及一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法。
背景技術:
喜樹堿(camptothecin)是在珙桐科落葉植物(nyssaceae)喜樹的根皮,或者種子中提取出來的,它是一個具有顯著的抵御腫瘤活體特性的堿性物質。目前,這是獨一被檢測到的通過遏制拓撲異構酶施展細胞毒性的一種自然植物活性因素。盡管喜樹堿存在著毒性和副作用,專業人士對于它及其衍生物的探尋的腳步從未停下,基于喜樹堿的構造和抗癌活性的關系很貼近。喜樹堿為淡黃色的針狀結構結晶體,能溶解在甲醇、naoh溶液和乙醇溶液中,但在水中卻不能溶解。其熔點為264~267℃,光照易變質,有輕微的吸濕性,遇到濃硫酸會顯黃綠色,用無菌水稀釋生成黃綠色熒光且溶液顯堿性。喜樹堿是五個環狀結構,a環是哇琳環,b環為哇琳環,c、d環分別為為毗咯環和毗咤酮結構構造,e環則是一個α—羥基內脂(包含有s型手性碳結構)。喜樹堿:c20h16n2o4;分子量348.3,分子結構如式1-1所示。
由于喜樹堿在自然界中有很多的衍生物,其中的一種名稱為cpt—11(1c)的藥物,其抑制癌癥有特別的機制而且在抑制或行進的途中不用依靠煙酰胺腺嘌呤二核苷酸和三磷酸腺苷酶等能量協助因素,所以很多研究此類物質的人員對其探索充滿了預期和愿景。喜樹堿抗癌的機理如式1-2所示,是拓撲異構酶與cpt的相互作用的過程。
喜樹堿常用的檢測方法有薄層色譜法、高效液相色譜法和紫外分光光度法等,其中,薄層色譜法的檢測結果較為單一,只能夠檢測出樣品的存在與否,不能直接或間接的測定樣品的含量;高效液相色譜法能夠準確并直接測得樣品中喜樹堿的含量,并且色譜圖清晰易懂,體現了高效液相色譜儀的靈敏度高、分析速度快、分辨率高、樣品用量少、易回收等特點。但是此方法測量時間較長、儀器較為昂貴、所用流動相通常為有機溶劑,對操作者健康及環境都將產生一定的危害。而紫外分光光度法具有準確度高、分析速度快、操作簡單等優點,為喜樹堿的檢測提供了快速、精確、便捷的目的。但由于實際樣品中,喜樹堿含量很低,儀器靈敏度受限等,直接測定并不能達到分析要求,因此在測定前有必要采用樣品分離和富集方法,提高分析檢測的靈敏度。
磁性固相萃取(mspe)是目前應用較多的一種樣品的分離和富集方法。該方法是根據固相萃取理論,憑借材料本身特性而實現樣品分離和富集的一種方法。mspe可以直接將磁性吸附劑添加到樣品的溶液中,待測物可以被磁性吸附劑吸附在其分散的表面,隨后用洗脫液將待測物質從吸附劑上解吸下來,達到了樣品與吸附劑分離的目的。該方法具有樣品用量少、萃取時間短、操作簡單、干擾小等特點。相對于傳統固相萃取吸附劑,磁性固相萃取吸附劑在實驗過程中會避免消耗大量吸附解吸時間而進入了我們的視線當中。由此,開發出適合分離和富集喜樹堿的新型吸附材料是目前亟待解決的問題。
技術實現要素:
為了解決現有技術中存在的問題,本發明提供了一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,所述磁性固相萃取吸附材料為β-環糊精改性的fe3o4磁性納米材料,通過β-環糊精的疏水性空腔與檢測物喜樹堿的疏水性之間的主客體包結作用來實現喜樹堿的富集。
本發明提供的利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,所述磁性固相萃取吸附材料為β-環糊精改性的fe3o4磁性納米材料,具體富集檢測過程如下:
將含有喜樹堿的樣品溶液的ph值調節至10,之后倒入裝有β-環糊精改性的fe3o4磁性納米材料的容器中,所述樣品溶液和β-環糊精改性的fe3o4磁性納米材料按照1ml:0.6~1mg的比例投加,恒溫水浴振蕩15~20min,振蕩完畢后用磁鐵分離吸附材料;
向分離的吸附材料中加入堿性洗脫液,振蕩8~15min,完畢后取出,用磁鐵分離吸附材料,采用紫外分光光度法在366nm下檢測解吸液的吸光強度。
優選地,所述方法的線性范圍在0.05~10μg/ml;檢出限為0.041μg/ml,相對標準偏差為8.7%。
優選地,所述β-環糊精改性的fe3o4磁性納米材料通過水熱合成法制備,具體過程如下:將β-cd、fe2+和fe3+溶于一定量的naoh溶液中,在氮氣氣氛下,混合液攪拌至完全溶解,然后將混合液轉移到水熱反應釜中,于160~165℃下反應16~24h,反應完畢后,用磁鐵將固液分離,用水和乙醇反復洗滌,將產物于75~80℃下真空干燥,即得β-環糊精改性的fe3o4磁性納米材料,記為β-cd@fe3o4納米材料;所述β-cd、fe2+和fe3+摩爾比為0.01~0.015:1:2。
更優選地,所述naoh溶液的濃度為4.0mol/l。
更優選地,所述fe2+通過feso4·7h2o提供,所述fe3+通過fe2(so4)3或fecl3·6h2o提供。
與現有技術相比,本發明具有如下有益效果:
(1)喜樹堿獨特的藥用價值以及臨床作用,已成為一種熱門的研究物質,其檢測尤為重要,本發明利用β-環糊精的疏水性空腔與檢測物喜樹堿的疏水性之間的主客體包結作用來實現喜樹堿的富集,有效避免了毒害作用的有機溶劑的使用,樣品洗脫的過程變得簡單易操作,大倍數地富集作用可以使一些痕量化合物得以被檢測出。
(2)本發明采用水熱法制備β-環糊精改性的fe3o4納米材料,制備的納米粒子大小比較均勻,且在制備過程中可以有效避免n2保護不徹底而導致fe被氧化的問題,也除去了用β-cd二次改性fe3o4的繁瑣步驟。
(3)環糊精與fe3o4均是無污染、生物相容性好的材料,有利于抗癌藥物喜樹堿在復雜樣品基質中的檢測。
(4)磁性固相萃取分離方法較為簡便,可以利用磁鐵對fe3o4納米材料的吸附即可實現固液分離,方便收集洗脫液用于樣品檢測,有效避免了繁瑣的離心或過濾步驟。
附圖說明
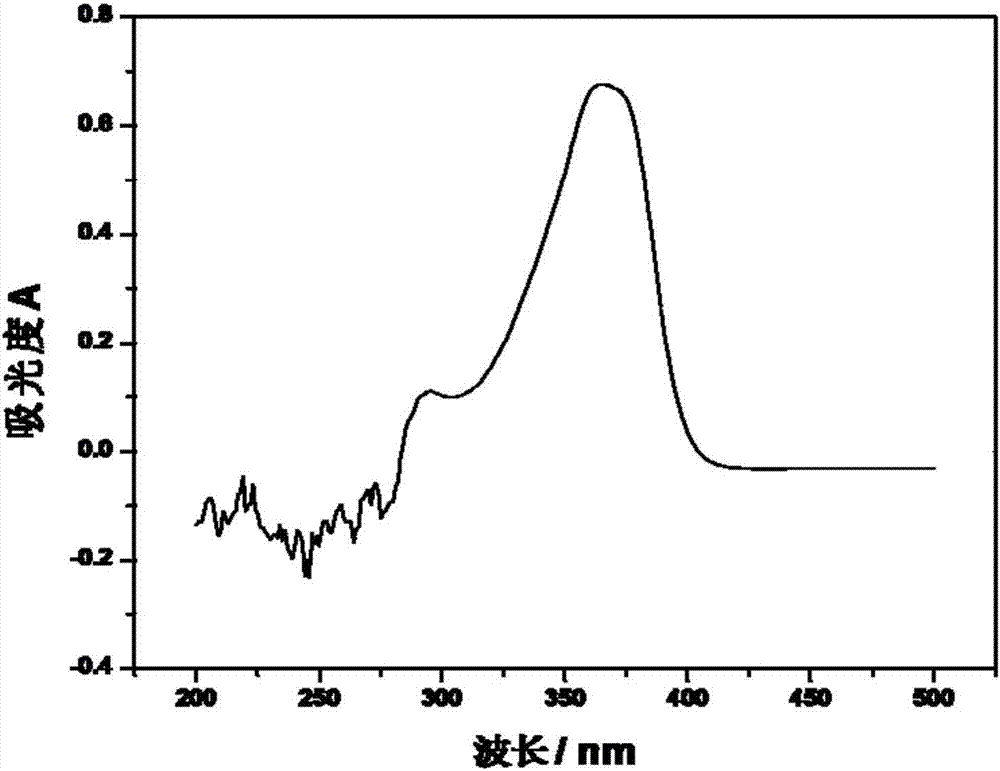
圖1是喜樹堿標準溶液的紫外光譜圖;
圖2是喜樹堿標準曲線;
圖3是本發明實施例中提供的β-cd@fe3o4磁性納米材料的紅外光譜圖;
圖4是本發明實施例中提供的fe3o4磁性納米材料的紅外光譜圖;
圖5是本發明實施例1、對比例1、對比例2對喜樹堿的檢測結果;
圖6是本發明實施例1、以及對比例3-5對喜樹堿的檢測結果。
具體實施方式
為了使本領域技術人員更好地理解本發明的技術方案能予以實施,下面結合附圖和具體實施例對本發明作進一步說明,但所舉實施例不作為對本發明的限定。
一、喜樹堿標準溶液的標定
我們配制了20μg/ml的喜樹堿標準溶液,并過該標準溶液進行標定,具體過程如下:稱取0.002g喜樹堿標準品于燒杯中,加入適量濃度的naoh水溶液,攪拌至其完全溶解,轉移到容量瓶中,得到20μg/ml的喜樹堿標準溶液,其紫外譜圖如圖1所示,由圖1可以看出其最大吸收波長為366nm。
然后我們將上述標準溶液逐級稀釋可得一系列濃度(0.05μg/ml、0.5μg/ml、1μg/ml、5μg/ml、10μg/ml)的喜樹堿溶液,分別利用紫外可見分光光度法在366nm下依次測定每個濃度的對應的吸光度,其吸光度的值見下表1所示。
表1喜樹堿不同濃度標準溶液吸光度
我們以濃度為橫坐標,吸光強度為縱坐標,用origin軟件繪制出濃度與吸光度的關系圖,即可得到喜樹堿標準曲線,具體如圖1所示。經線性擬合得標準曲線方程為:y=0.05283x-0.00432r2=0.99958,說明其線性關系良好。
二、β-cd@fe3o4磁性納米材料的制備
采用水熱合成法制備β-cd@fe3o4磁性納米材料,具體過程如下:取12.8gβ-cd,溶于4mol/lnaoh溶液中至完全溶解,再加入2.78gfeso4·7h2o和4gfe2(so4)3,將此混合溶液n2保護下攪拌至完全溶解。然后轉移到聚四氟乙烯反應釜中,在160℃下反應24h。反應完畢后,用磁鐵將固液分離,用水和乙醇反復洗滌,將產物放在80℃下真空干燥。
我們對上述過程制備得到的β-cd@fe3o4磁性納米材料進行表征,具體稱取約2mg的β-cd@fe3o4樣品進行研磨,以kbr為背景,通過紅外光譜儀對該化合物進行紅外光譜的掃描,得到的紅外光譜圖具體圖圖1所示。
通過圖3可以看出fe3o4的fe-o的特征吸收峰呈現在580cm-1處,此處較為平坦可能是因為制備過程中微量fe氧化導致;1630cm-1處是β-cd分子中面內彎曲振動峰,且3400cm-1出現了一個明顯的特征吸收峰,可能是β-cd@fe3o4納米材料表面的-oh氫氧鍵伸縮振動吸收峰,因此可以推斷出本實施例成功制備了β-cd@fe3o4納米材料。
三、fe3o4磁性納米材料的制備
具體過程如下:50ml三口燒瓶中加入20ml蒸餾水,機械攪拌,通n2除氧約20-30min,然后加入1.08gfecl3·6h2o,0.556gfeso4·7h2o,繼續攪拌10min(攪拌速度為500r/min),溶液呈橙紅色,水浴加熱到60℃,緩慢滴加2.5ml濃氨水到上述溶液中,滴加時間大概為1min,溶液的ph在9-10之間,溶液迅速由橙紅色變為黑色,繼續攪拌反應20min后升高到80℃并停止攪拌,放置20min,將8ml上述溶液移至10ml水熱釜中,將水熱釜置于140℃的烘箱中,水熱反應6h,反應結束后,將水熱釜冷卻至室溫,得到亮黑色沉淀。烘干即可得到fe3o4納米粒子。
我們對上述過程制備得到的fe3o4磁性納米材料進行表征,具體稱取約2mg的fe3o4樣品進行研磨,以kbr為背景,通過紅外光譜儀對該化合物進行紅外光譜的掃描,得到的紅外光譜圖具體圖圖4所示。
通過圖4可以看出fe3o4的fe-o的特征吸收峰呈現在580cm-1處,可以看出fe3o4的fe-o的特征峰在580cm-1處;在700cm-1也可以判斷出不存在亞鐵氧鍵的特征吸收峰,所以不存在氧化鐵物質;-oh氫氧鍵伸縮振動吸收峰在3320cm-1處,表明了fe3o4納米粒子的表面羥基。
實施例1
本實施例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,所述磁性固相萃取吸附材料為上述水熱合成法制備的β-cd@fe3o4磁性納米材料;具體富集檢測過程如下:將20ml的1μg/ml喜樹堿樣品的ph值調節至10,倒入裝有20mgfe3o4@β-cd吸附材料的錐形瓶中,恒溫水浴振蕩20min。振蕩完畢后取出錐形瓶,用磁鐵分離吸附劑后將上清液除去,加入5ml洗脫液1mol/lnh3·h2o振蕩10min,完畢后取出,用磁鐵分離吸附劑,收集上清液,采用紫外分光光度法在366nm下檢測解吸液的吸光強度,重復3次,取平均值。
對比例1
本對比例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,所述磁性固相萃取吸附材料為上述制備的fe3o4磁性納米材料;具體富集檢測過程和實施例1相同。
對比例2
本對比例一種檢測喜樹堿的方法,直接對20ml的1μg/ml喜樹堿樣品采用紫外分光光度法在366nm下測定其吸光強度。
對比例3
本對比例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,具體富集檢測過程和實施例1相同,不同之處僅在于,洗脫劑選擇5ml的2mol/lnah2po4。
對比例4
本對比例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,具體富集檢測過程和實施例1相同,不同之處僅在于,洗脫劑選擇5ml的甲醇:水=6:4(v/v)。
對比例5
本對比例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,具體富集檢測過程和實施例1相同,不同之處僅在于,洗脫劑選擇5ml的乙腈:水=6:4(v/v)。
對比例6
本對比例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,具體富集檢測過程和實施例1相同,不同之處僅在于,將20ml的1μg/ml喜樹堿樣品的ph值調節至11。
對比例7
本對比例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,具體富集檢測過程和實施例1相同,不同之處僅在于,將20ml的1μg/ml喜樹堿樣品的ph值調節至7。
對比例8
本對比例一種利用磁性固相萃取吸附材料富集檢測喜樹堿的方法,具體富集檢測過程和實施例1相同,不同之處僅在于,將20ml的1μg/ml喜樹堿樣品的ph值調節至6。
實施例1使用β-cd@fe3o4磁性納米材料的富集性能與對比例1的fe3o4磁性納米材料fe3o4磁性納米材料和對比例2直接紫外測試喜樹堿樣品的檢測結果如圖5所示,由圖5可以看出,經β-cd@fe3o4磁性納米材料富集后的吸光度明顯高于其他兩種分析方法,這是由于β-cd@fe3o4納米材料與喜樹堿之間存在弱的主客體相互作用,一定程度上提高了喜樹堿的富集能力,富集更為完全,檢測結果更為精確。表明β-cd@fe3o4磁性納米材料對喜樹堿有更高的富集性能。
進一步地,實施例1、以及對比例3-5分別選用了不同的洗脫劑,他們對喜樹堿樣品的檢測結果如圖6所示,由圖6可以明顯看出,用1mol/lnh3·h2o作洗脫液時,吸光度值最大,說明nh3·h2o對喜樹堿的洗脫效果更好,可以最大限度地將吸附在吸附劑的喜樹堿洗脫到溶液中。其原因可能是因為nh3·h2o是一種堿性物質,可以破壞喜樹堿與β-cd的主客體分子間作用力,從而達到洗脫的目的。
進一步地,實施例1中喜樹堿樣品的ph值是10,對比例6中喜樹堿樣品的ph值是11,對比例7中喜樹堿樣品的ph值是7,對比例8中喜樹堿樣品的ph值是6,其他條件均相同。他們對喜樹堿樣品的檢測結果可知,當喜樹堿樣品溶液的ph為10時,喜樹堿對于富集和萃取的性能優化效果最好,達99%左右;當喜樹堿樣品溶液的ph為11時,為60.7%;當喜樹堿樣品溶液的ph為7時,為68.2%;當喜樹堿樣品溶液的ph為6時,為40%;可見堿性條件下其富集效果較佳,其中以ph為10為高,而酸性條件下富集效果差;這是由于喜樹堿在ph小于4.5時,分子結構以內酯環形式存在;ph大于7.4時,分子結構以開環羧酸鹽形式存在;在堿性條件下,其開環結構使得其更容易被吸附劑吸附,從而富集效果更佳。
進一步地,我們對實施例1提供的方法進行準確度評價,檢出限和定量限分別以信噪比s/n的3倍和10倍計算,測得結果分別為0.041μg/ml和0.137μg/ml,精密度8.7%。表明該方法準確、可靠,適用于喜樹堿的檢測。
以上所述實施例僅是為充分說明本發明而所舉的較佳的實施例,其保護范圍不限于此。本技術領域的技術人員在本發明基礎上所作的等同替代或變換,均在本發明的保護范圍之內,本發明的保護范圍以權利要求書為準。
- 還沒有人留言評論。精彩留言會獲得點贊!