一種CO脫氫凈化用Pd?Cl/CeOCl催化劑及制備方法與流程
本發明涉及催化劑的制備技術,具體涉及一種pd-cl/ceocl催化劑的制備方法,應用于co原料氣的脫氫凈化反應。
背景技術:
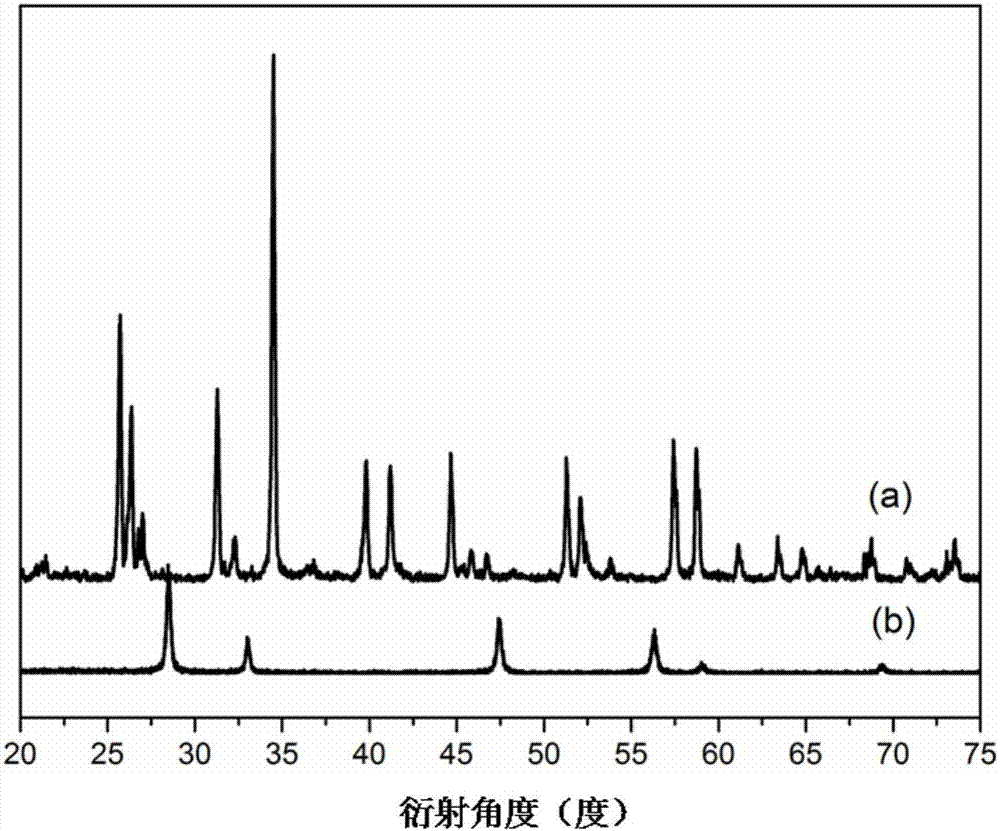
:co是合成草酸酯、碳酸酯等有機化學品的重要原料。通常由煤、甲烷、生物質等化石資源重整得到合成氣(co+h2),再經過變壓(變溫)吸附分離得到co原料氣。這些原料氣含有0.5~5%的h2雜質,嚴重影響co合成草酸酯或碳酸酯的效率,需要通過選擇氧化反應予以脫除凈化。該反應通常由負載型貴金屬(pd、pt、rh)催化劑驅動,催化劑的穩定性是制約脫氫反應的關鍵所在。中國專利201410309019.4曾公開一種高性能pd-cl/al2o3脫氫催化劑、中國專利201610520443.2曾公開一種缺陷型pdclx/al2o3脫氫催化劑,通過修飾pd活性中心的電子結構,該系列催化劑具備很高的脫氫活性和選擇性。然而,由于co誘導的奧斯瓦爾德熟化效應會加速活性金屬的燒結聚集、反應產物h2o的侵蝕會破壞活性中心的結構,經過上千小時反應后催化劑的性能會不可避免的下降。針對以上問題,本發明制備一種具有高穩定性的pd-cl/ceocl催化劑,利用pd-cl-ce單體中cl的橋連作用增強pd活性組分與ceocl載體間的強相互作用。該催化劑對co和h2o的侵蝕皆具有良好的免疫性,在反應環境下的穩定性顯著提高。技術實現要素:本發明旨在提供一種高穩定性pd-cl基催化劑及其制備方法,該催化劑主要應用于草酸酯或碳酸酯合成用高純co原料氣中h2雜質的選擇氧化脫除。本發明所提供的催化劑的化學表示式為:pd-cl/ceocl,催化劑中單質pd占催化劑的質量百分數為0.5~2wt.%;催化劑中cl/pd的摩爾比為3~2:1;催化劑載體為具有四方晶相的ceocl。該催化劑以含氯鈀鹽作為活性組分前體,以ceo2作為載體前體,通過精細控制的原位復合過程在活性組分—載體的界面上形成具有高穩定性的pd-cl-ce單體,在此過程中ceo2前體轉化為ceocl,同時pd-cl的鍵能及其與ceocl載體的相互作用顯著增強。上述催化劑的制備方法,具體步驟如下:a.稱取含氯鈀鹽溶于無水乙醇中,再按表面活性劑與pd的摩爾比為1:10~20加入表面活性劑,超聲5~6h,配制濃度為0.01~0.04mol/l的活性組分溶液a;所述含氯鈀鹽為氯化鈀或氯鈀酸銨,優選為氯化鈀;所述表面活性劑為油酸、聚乙烯吡咯烷酮、聚合度為200~600的聚乙二醇中的一種,優選為聚乙烯吡咯烷酮。b.將ceo2干溶膠分散于無水乙醇中,再加入表面活性劑,超聲0.5~1h得到濃度為0.3~0.6mol/l的溶液b;其中表面活性劑與ce的摩爾比為1:1~5;所述表面活性劑為氯化銨、十四烷基三甲基氯化銨、十六烷基三甲基氯化銨的其中之一,優選為十六烷基三甲基氯化銨。c.在惰性氣體保護下,將溶液b緩慢滴加入溶液a中,其中a、b溶液的加入量按照最終催化劑中pd占催化劑的質量百分數確定,并保持1000~3000rpm的攪拌速度,于20~60℃反應4~12h后,于0.02-0.03mpa真空干燥10~12h,得到固體樣品。d.將步驟c得到的固體樣品置于石英反應管中,在惰性氣體保護下于300~500℃處理4~8h后降至室溫,再通入h2濃度為5~20%的h2/ar混合氣以1~3℃/min的速率升溫至500~650℃并穩定6~8h后降至室溫,在室溫下通入惰性氣體鈍化處理10~12h后得到pd-cl/ceocl催化劑成品。圖1為實施例1中pd-cl/ceocl催化劑的高分辨透射電鏡照片,插圖為ceocl的傅里葉變換衍射圖,通過晶面間距和衍射斑排列證明在催化劑的制備過程中ceo2完全轉化為ceocl。圖2為催化劑樣品的x射線粉末衍射譜圖:(a)表示實施例1的pd-cl/ceocl催化劑,(b)表示比較例1的pd-cl/ceo2催化劑,通過衍射峰的位置證明在催化劑的制備過程中ceo2完全轉化為ceocl。本發明的有益效果體現在:通過精細控制催化劑活性組分與載體的原位復合過程,以及后處理過程中的還原速度、溫度、鈍化時間等工藝參數,誘導了載體由ceo2向ceocl的轉化并促進了pd-cl-ce單體的形成,在強化了pd-cl鍵的同時還增強了活性組分—載體間的相互作用。與現有脫氫催化劑相比,本發明制備得到的催化劑對co誘導的奧斯特瓦爾德熟化效應、產物h2o的侵蝕皆具備良好的免疫性,在反應氣氛下具有更高的穩定性,具有超過1000小時的壽命。附圖說明圖1為實施例1中pd-cl/ceocl催化劑的高分辨透射電鏡照片,插圖為ceocl的傅里葉變換衍射圖。圖2為兩個催化劑樣品的x射線粉末衍射譜圖:(a)為實施例1的pd-cl/ceocl催化劑,(b)為比較例1的pd-cl/ceo2催化劑。具體實施方式實施例1:a.稱取0.089g氯化鈀溶于50ml無水乙醇中,加入聚乙烯吡咯烷酮,超聲處理5h后配制得到濃度為0.01mol/l的活性組分溶液。b.稱取2.7gceo2干溶膠分散于25ml無水乙醇中,加入十六烷基三甲基氯化銨,超聲處理0.5h得到濃度為0.6mo/l的溶液。c.將步驟b得到溶液緩慢滴加入步驟a得到的溶液中,期間始終用惰性氣體保護并保持3000rpm的攪拌速度。于20℃反應10h后,于0.02mpa真空干燥10h,得到固體樣品。d.將步驟c得到的固體樣品置于石英反應管中,在惰性氣體保護下于300℃處理8h后降至室溫,再通入h2濃度為20%的h2/ar混合氣以1℃/min的速率升溫至650℃并穩定6h后降至室溫,在室溫下通入惰性氣體處理10h后得到催化劑pd-cl/ceocl。通過等離子發射光譜測得該催化劑中pd占催化劑的質量百分數為0.63wt.%;通過光電子能譜測得催化劑中cl/pd的表面濃度比為2.2:1。取1g催化劑進行1000h穩定性測試考察其應用性能,具體測試條件是:反應溫度140℃,反應壓力0.25mpa,原料氣空速2000h-1,待處理原料氣配比為co占98.5%,h2占1.5%,加入o2與h2的流量比為2:1,催化劑性能評價結果見表1。采用co靜態化學吸附法分析上述運行1000h后的催化劑的表面pd分散度,采用x射線精細結構分析其活性中心的pd-cl配位結構,結果見表2。實施例2:同實施例1的制備過程,區別在于步驟a中氯化鈀的加入量為0.36g,活性組分溶液濃度為0.04mol/l。通過等離子發射光譜測得該催化劑中pd占催化劑的質量百分數為1.99wt.%;通過光電子能譜測得催化劑中cl/pd的表面濃度比為2.2:1。同實施例1的催化劑評價條件,性能評價結果見表1。反應后催化劑表面pd物種的分散度和pd-cl配位結構分析結果見表2。實施例3:a.稱取0.089g氯化鈀溶于50ml無水乙醇中,加入聚乙烯吡咯烷酮,超聲處理5h后配制得到濃度為0.01mol/l的活性組分溶液。b.稱取2.7gceo2干溶膠分散于25ml無水乙醇中,加入氯化銨,超聲處理0.5h得到濃度為0.6mo/l的溶液。c.將步驟b得到溶液緩慢滴加入步驟a得到的溶液中,期間始終用惰性氣體保護并保持1000rpm的攪拌速度。于60℃反應4h后,于0.02mpa真空干燥10h,得到固體樣品。d.將步驟c得到的固體樣品置于石英反應管中,在惰性氣體保護下于300℃處理8h后降至室溫,再通入h2濃度為5%的h2/ar混合氣以3℃/min的速率升溫至500℃并穩定6h后降至室溫,在室溫下通入惰性氣體處理10h后得到催化劑pd-cl/ceocl。通過等離子發射光譜測得該催化劑中pd占催化劑的質量百分數為0.64wt.%;通過光電子能譜測得催化劑中cl/pd的表面濃度比為2.8:1。同實施例1的催化劑評價條件,性能評價結果見表1。分析反應后催化劑表面pd物種的分散度和pd-cl配位結構,結果見表2。實施例4:同實施例3的制備過程,區別在于步驟a中加入的鈀鹽為0.14g氯鈀酸銨。通過等離子發射光譜測得該催化劑中pd占催化劑的質量百分數為0.61wt.%;通過光電子能譜測得催化劑中cl/pd的表面濃度比為3:1。同實施例1的催化劑評價條件,性能評價結果見表1。分析反應后催化劑表面pd物種的分散度和pd-cl配位結構,結果見表2。對比例1:按照中國專利201410309019.4公開的實施例2中的制備方法得到pd-cl/al2o3催化劑,將其作為對比例。同本發明實施例1的評價條件進行催化劑穩定性測試,性能評價結果見表1。分析反應后催化劑表面pd物種的分散度和pd-cl配位結構,結果見表2。表1:催化劑1000h后的性能評價結果脫氫后h2濃度(ppm)h2選擇性(%)實施例16581.4實施例28083.3實施例310178實施例49978.9對比例171757.1表1數據表明,本發明制備的催化劑的脫氫效率及脫氫穩定性遠優于對比例1的pd-cl/al2o3催化劑。表2:反應1000h后催化劑中pd物種的分散度及pd-cl配位結構表2數據表明:本發明制備的催化劑結果1000h運行后仍保持良好的pd散度和初始pd-cl活性中心結構,遠優于對比例1的pd-cl/al2o3催化劑。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1