一種用于甲醇氫氯化合成一氯甲烷的催化劑的制備方法與流程
本發明屬于一氯甲烷合成領域,具體涉及一種用于甲醇氫氯化合成一氯甲烷的催化劑的制備方法。
背景技術:
一氯甲烷俗稱氯甲烷,是使用量較大的化學中間體和化工原料,主要用于制備有機硅行業的原料,甲基氯硅烷單體、也用于甲基纖維素、季胺化合物、農藥、有機化合物的溶劑和乳化劑等的生產,還可用作醫藥麻醉劑。甲烷氯化物的中間原料一氯甲烷是比甲烷更活潑的物質,可以作為中間產物合成下游的產品,其作用可與煤化工行業的甲醇相類似。近年來隨著有機硅、甲烷氯化物行業的迅猛發展,一氯甲烷的需求量也不斷增加。一氯甲烷的制備方法主要有:甲烷氯化法和甲醇氯化法。目前國內一氯甲烷生產方法以甲醇氯化法(甲醇液相催化法和甲醇氣相氯化法)為主。液相催化法是將甲醇和氯化氫混合進入裝有液相催化劑的反應釜進行液相反應。氣相甲醇氫氯化法是將氣體甲醇與氯化氫在裝有催化劑的固定床反應器內進行反應。
目前甲醇氫氯化合成一氯甲烷主要以商品
技術實現要素:
本發明要解決的技術問題是克服現有的缺陷,提供了一種用于甲醇氫氯化合成一氯甲烷的催化劑的制備方法。
為了解決上述技術問題,本發明提供了如下的技術方案:
一種用于甲醇氫氯化合成一氯甲烷的催化劑的制備方法,包括將
優選地,所述氯化鋅溶液的溶劑為有機溶劑,采用有機溶劑可有效抑制氯化鋅的水解。
更優選地,所述有機溶劑為甲醇、乙醇、甘油、丙酮或乙醚。
優選地,烘干溫度為30~80℃,焙燒溫度為300~600℃。更優選地,烘干溫度為50℃,焙燒溫度為400℃。優選地,焙燒的升溫速率為1~10℃/min,焙燒時間為2-4小時。
優選地,所述催化劑中zncl2的負載量為8wt%~15wt%。
更優選地,所述催化劑中zncl2的負載量為11wt%。
附圖說明
附圖用來提供對本發明的進一步理解,并且構成說明書的一部分,與本發明的實施例一起用于解釋本發明,并不構成對本發明的限制。在附圖中:
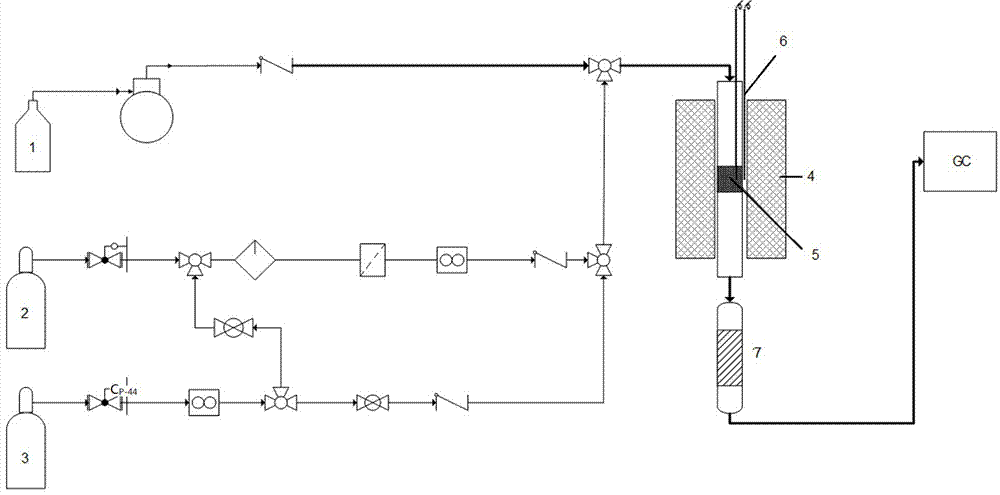
圖1是本發明催化劑性能評價實驗裝置示意圖,其中,1.甲醇,2.氯化氫,3.混合氣ch4/n2,4.加熱爐,5.催化劑,6.熱電偶,7.中和瓶。
具體實施方式
以下結合附圖對本發明的優選實施例進行說明,應當理解,此處所描述的優選實施例僅用于說明和解釋本發明,并不用于限定本發明。
實施例1
將異丙醇鋁粉末充分研磨至細粉末,稱取17.1g蔗糖溶于90ml的去離子水中,攪拌,使其充分溶解,緩慢加入10.2g磨細的異丙醇鋁,強攪拌使其溶解。調節ph至5.5,攪拌48h,80℃揮發易揮發組分,得到透明凝膠,以3℃/min的升溫速率升溫至600℃,焙燒4h,得到
稱取0.136gzncl2溶解至2.5ml去離子水中,加入0.1ml65w%的濃硝酸,使其充分溶解,加入1.02g上述制備的
實施例2
稱取0.136gzncl2溶解至4ml無水乙醇中,充分溶解,加入1.02g
實施例3
稱取0.0907gzncl2溶解至4ml無水乙醇中,充分溶解,加入1.02g
實施例4
稱取0.1813gzncl2溶解至4ml無水乙醇中,充分溶解,加入1.02g
對比例1
將異丙醇鋁粉末充分研磨至細粉末,稱取17.1g蔗糖溶于90ml的去離子水中,攪拌,使其充分溶解,緩慢加入10.2g磨細的異丙醇鋁,強攪拌使其溶解。然后加入0.34gzncl2,強攪拌3h,調節ph至5.5,攪拌48h,在80℃揮發易揮發組分,得到透明凝膠,以3℃/min的升溫速率升溫至600℃,焙燒4h,得到氯化鋅摻雜量為11wt%催化劑。
對比例2
稱取0.136gzncl2溶解至2.5ml去離子水中,充分溶解,加入1.02g
對比例3
稱取0.136gzncl2溶解至2.5ml去離子水中,充分溶解,加入1.02gγ-al2o3,在50℃烘干(空氣氣氛),以3℃/min的速率升溫至400℃,焙燒180min,得到氯化鋅負載量為11wt%催化劑。
催化劑性能評價實驗:將上述制備的
上述結果表明,采用本發明的方法可明顯改善zncl2/
本發明并不限于采用上述方法制備的
最后應說明的是:以上所述僅為本發明的優選實施例而已,并不用于限制本發明,盡管參照前述實施例對本發明進行了詳細的說明,對于本領域的技術人員來說,其依然可以對前述各實施例所記載的技術方案進行修改,或者對其中部分技術特征進行等同替換。凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!