復合型堿性鐵鉬硫化物催化劑及制備方法及其在芳香酚及醚類轉化中的應用與流程
本發明屬于工業催化和生物能源轉化技術領域,涉及到金屬硫化物催化劑的制備,具體地說是一種復合型堿性鐵鉬硫化物催化劑制備方法。
背景技術:
隨著人類對環境污染和資源危機等問題的認識不斷深入,天然高分子所具有的可再生、可降解等性質日益受到重視。而生物質作為一種天然的高分子,相比于化石能源,不僅具有儲備豐富,碳中性,易獲取,零排放,而且是唯一可轉化為液體燃料的可再生資源等眾多突出的優點。其中,木質生物質是生物質能源的重要組成部分,其主要由纖維素(40‐50%)、半纖維素(25‐35%)及木質素(15‐20%)等有機物組成。由于木質素復雜的結構以及難以降解的特點,通常通過催化加氫對木質素進行的高值化利用。但是其原料的處理仍然是個難點,目前主要通過研究其模型化合物即芳香酚轉化而進一步揭示其木質素原料的的轉化規律。
催化加氫是轉化木質素最常用的手段之一。根據產物的不同,目前對木質素模型化合物的轉化反應主要分為兩類。一是實現芳香結構的完全加氫生成環烷烴,用于生物油的制備;二是對其進行脫氧而保留芳香結構,用于高附加值化學品的制備。生物油的精煉技術發展至今,已經取得了很多矚目的成果,而在精細化學品的制取上,遠遠不夠。因此,如何提高目前催化劑體系對催化獲得酚類產物的選擇性是迫在眉睫的問題。
在目前應用的催化劑體系中,硫化物催化劑表現出突出的優勢。相比于貴金屬pd,pt以及ni金屬等催化劑,硫化物催化劑不僅不會造成催化劑中毒,反而有助于活性的穩定。最重要的是對酚類產物的選擇性上大大的優于金屬類催化劑,這有利于我們制取高附加值的酚類化學品。傳統的硫化物催化劑主要是指al2o3負載的como‐s或者nimo‐s催化劑,該類催化劑廣泛應用于工業當中,但是其活性遠遠沒有達到工業的需求,仍然需要進一步的提高。除此之外,鐵硫化合物也表現出了優異的加氫脫氧能力。ji等人通過浸漬法合成了負載型的二硫化亞鐵催化劑,在二芐醚的轉化中達到100%轉化,且對脫氧產物甲苯的選擇性達到99%,但是該催化劑需要在無氧條件下制備,且需要的壓力為10mpa限制了它的應用;此外,二芐醚中的c‐o鍵斷裂需要的鍵能較低,其在其它分類物質中的轉化效果未知。(najietal,iron(ii)disulfidesasprecursorsofhighlyselectivecatalystsforhydrodeoxygenationofdibenzyletherintotoluene,chemcatchem2015,7,960–966)。
綜上所述,除了傳統的硫化物催化劑,新的元素也在逐漸的開發當中。然而另一方面,傳統的硫化物也必然存在其優勢,而如何將兩者結合起來,具有一定的挑戰性。
技術實現要素:
為了解決現有技術的問題,本發明提出一種復復合型堿性鐵鉬硫化物催化劑及制備方法及其在芳香酚及醚類轉化中的應用。
本發明采取的技術方案為:
一種復合型堿性鐵鉬硫化物催化劑,其催化劑用分子式mgxfey‐mo‐s表示,x,y代表的是原子比,范圍為x:y=3:1‐1:3。
本發明的復合型堿性鐵鉬硫化物催化劑的制備方法;以mgxfey水滑石為前體,350‐500℃焙燒后得到mgxfey的氧化物,然后通過與含過量moo42‐離子的水溶液通過水合得到插層的mgxfey‐moo42前體,通過h2s/h2混合氣在400‐800℃下硫化,最終得到mgxfey‐mo‐s催化劑。
所述h2s/h2混合氣的體積比例為h2s:混合氣=10‐60vol%。
所述硫化時間為2‐10小時。
本發明的催化劑在芳香酚及醚類轉化中應用,將反應物溶于有機溶劑中,形成濃度為0.1‐0.5mol/l的反應液,按質量比反應物:催化劑=10:1‐5:1稱取催化劑,然后連同反應液和催化劑儀器裝入反應釜中,密閉后,通入氫氣置換3‐5次,確保完全除去釜中的空氣后,充入氫氣至反應壓力為1‐6mpa,攪拌速率為100‐2000r/min攪拌同時加熱升溫至反應溫度150‐350℃,反應1‐6h時間,降至室溫,取液體產物。
所述酚類及醚類化合物為丁香酚、愈創木酚、苯甲醚、二苯醚、二芐醚或芐基苯基醚中的一種。
所述有機溶劑為甲基環己烷、環己烷、十氫化萘、四氫呋喃或1,4‐二氧六環中的一種。
優選加熱升溫反應溫度范圍為300-350℃,反應時間為3h–4h。
優選攪拌速率為800‐1200r/min。
優選室溫下反應釜中氫氣的初始壓力4‐5mpa。
本發明中最終產物的分析方法:用氣相色譜‐質譜聯用儀和氣相色譜儀分別對得到的液體產物進行定性和定量檢測。以十二烷為內標物,通過內標法做出各產物的標準曲線,根據標準曲線計算,最終得到各種產物的濃度分布,進而計算轉化率及產率。轉化率以(反應物初始摩爾濃度‐反應物剩余摩爾濃度)/(反應物初始摩爾濃度)x100%計算。產物的產率以(產物摩爾數/nc)/(反應物初始摩爾數)x100%進行計算(nc代表產物與反應物中6元環的數量的比值)。
從目前文獻的調研結果看,尚未有任何報道是以mgfe水滑石為前體,通過離子水合引入活性組分mo元素,最終硫化得到以fe1‐xs及mos2為主要加氫活性組分,mgo提供堿性位的復合型催化劑,用于芳香酚及醚類的催化轉化。一方面考察催化劑在該反應體系中的活性,另一方面通過加氫脫氧反應,實現反應物中芳醚鍵的斷裂,得到含氧量低的酚類產物,提高其利用附加值。
本發明有如下優點:
1.本發明以mgfe水滑石為模板,有利于mos2的分散,提高其活性;
2.獲得的催化劑中包涵mos2和fe1‐xs兩種活性中心,分飾著不同的作用,而且兩者存在協同作用;
3.本發明復合了堿性載體和鐵鉬硫化物,簡便的制備了多功能的體相催化劑
附圖說明
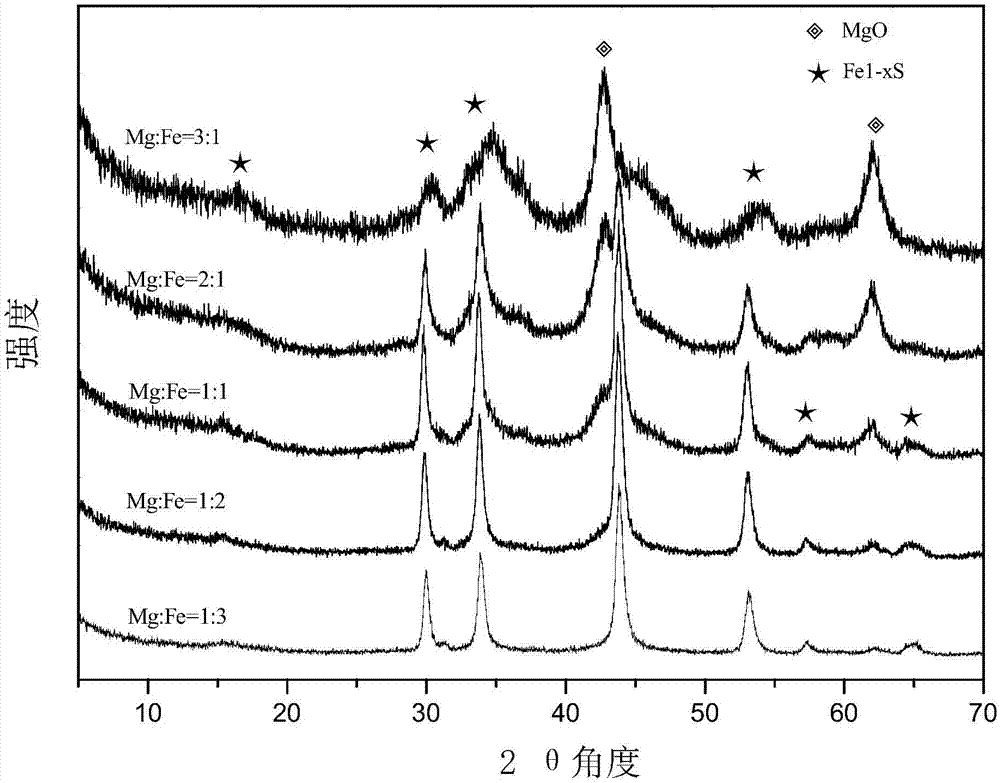
圖1為不同比例(mg:fe=3:1‐1:3)獲得的水滑石的xrd圖;
圖2為不同比例(mg:fe=3:1‐1:3)獲得的復合型堿性鐵鉬硫化物催化劑的xrd圖。
具體實施方式
下面通過具體實施例予以進一步的詳細說明。
實施例1
本實施例說明本發明中不同比例的水滑石的制備,以mg:fe=3:1為例
(a)稱量21.77g(0.09mol)硝酸鎂,12.12g(0.03mol)硝酸鐵溶于250ml去離子水中,攪拌至澄清;稱量9.6g(0.24mol)氫氧化鈉,6.36g(0.06mol)碳酸鈉溶于250mol去離子水中,攪拌至澄清;
(b)將兩種溶液同時滴入500ml去離子水中,70℃水浴攪拌,控制滴定的速度保證ph=9‐10之間;滴定完后,繼續攪拌,保持120分鐘,然后轉移到烘箱中,晶化溫度設定在70℃,晶化時間為12小時。
(c)晶化結束后用去離子水過濾洗滌沉淀,至ph為中性,所得沉淀在70℃下干燥12小時,即獲得原子比為mg:fe=3:1的mg3fe1水滑石。
其余比例的mgxfey水滑石的制備過程類似,不同之處在于改變硝酸鎂和硝酸鐵的量,保證n(mg)+n(fe)=0.12mol,其余條件不變;
具體合成的水滑石的xrd見圖1所示,隨著fe含量的增加,水滑石典型的層狀結構逐漸崩塌,說明fe的加入,使得形成的水滑石結構逐漸崩塌,將影響最終獲得的催化劑的層狀結構保留程度。
實施例2
本實施例說明本發明中水滑石基復合氧化物的制備
將實施例1制備得到的不同比例的水滑石置于馬弗爐中,設置升溫程序為5‐20k/min,升至400℃,得到該溫度下的不同比例mgxfey氧化物。
改變焙燒的溫度為350‐500℃之間,保證co32‐的去除而同時又保持其結構的可恢復性。
實施例3
本實施例說明本發明中復合硫化物催化劑的制備
(a)稱取2.0g七鉬酸銨溶于16mol去離子水中,攪拌形成澄清溶液,然后稱取1.0g實施例3中400℃煅燒制得的mgxfey氧化物加入到溶液中,用封口膜密封燒杯口,室溫下離子交換12小時,洗滌干燥得到mgxfey-moo42-前體;然后在10%的h2s/h2混合氣中硫化,硫化溫度為400℃,時間為4小時,最終獲得堿金屬復合的鐵鉬硫化物催化劑mgxfey-mo-s。根據比例的不同,分別記為mg3fe1mo-s,mg2fe1mo-s,mg1fe1mo-s,mg1fe2mo-s,mg1fe3mo-s.
該條件下制備的催化劑的xrd見圖2所示,最終并沒有觀察到mos2的衍射峰,說明mos2高度分散。另外,mg主要以堿性mgo的形式存在,為催化劑提供堿性位,fe主要以fe1‐xs的形式存在,隨著fe比例的增加,催化劑的主要晶相由mgo變為fe1‐xs。
改變h2s/h2混合氣的體積比為20%,30%,40%,50%,60%,獲得不同硫化氣氛濃度下mgxfey‐mo‐s。
改變硫化過程的溫度450℃,500℃,550℃,600℃,650℃,700℃,750℃,800℃,獲得不同硫化溫度下mgxfey‐mo‐s。
改變硫化時間為2,3,5,6,7,8,9,10小時,獲得不同硫化時間下mgxfey‐mo‐s。
實施例4
本實施例將實施例3中不同比列的硫化物催化劑用于丁香酚加氫脫氧反應中,具體操作為:
將0.4105g(2.5mmol)丁香酚和0.4258g(2.5mmol)十二烷溶解甲基環己烷中至體積為20ml,連同0.0821gmgxfeymo‐s催化劑加入到50ml大小的間歇式反應釜中,通入氫氣置換五次氣體后,充氫氣至5mpa,以1000r/min的速度進行攪拌,同時升溫至300℃反應3h,反應時間從反應溫度達到300℃時開始計時。反應結束后,降至室溫,得到液體產物。分析結果如表1所示,五種不同比例的催化劑均對丁香酚表現出了很高的催化活性,其中當mg:fe=1:2時,催化劑的活性最高,達到了100%的丁香酚轉化,以及得到了44.0%的4‐丙基苯酚產率,以及超過60%產率的酚類產物。相比于同類應用于該反應物的催化劑,該催化劑表現出了突出的酚類產物選擇性,為工業制高附加值的精細化學品提供了新的選擇。
表1不同比例催化劑活性對比
實施例5
本實施例將考察反應溫度對催化劑活性的影響,以mg1fe2mo‐s為催化劑,丁香酚為反應物。實驗過程類似于實施例4,不同之處在于反應溫度的變化,反應結果見表2。
由表可知,在250℃下,丁香酚沒有完全轉化,當溫度達到300℃時,4‐丙基苯酚的產率達到最高。進一步升高反應溫度將導致4‐丙基苯酚的進一步轉化生成正丙苯。說明該催化體系下300℃為最優反應溫度。
表2不同反應溫度對比
實施例6
本實施例將考察不同氫氣初始壓力對催化劑活性的影響,以mg1fe2mo‐s為催化劑,丁香酚為反應物。實驗過程類似于實施例4,不同之處在于初始氫氣壓力的變化,結果見表3。
可以看出,隨著壓力的升高,丁香酚的轉化率逐漸升高,并且有利于酚類產物的生成。但是過高的壓力也會導致酚類產物的進一步氫解。當壓力為5mpa時,該反應的轉化率達到100%,酚類產物的產量也達到最高值。
表3不同反應初始氫壓對比
實施例7
本實施例將考察不同反應時間對催化劑活性的影響,以mg1fe2mo‐s為催化劑,丁香酚為反應物。實驗過程類似于實施例4,不同之處在于反應時間的變化,結果見表4。
隨著反應的進行,轉化率在3小時時達到100%,4‐丙基苯酚的產率先升高緩緩降低,說明反應的速控步驟為4‐丙基苯酚的進一步轉化。
表4不同反應時間對比
實施例8
本實施例將實施例上述mg1fe2mo‐s催化劑應用于4‐丙基苯酚的加氫脫氧反應中。實驗過程類似于實施例4,不同之處在于將反應物替換為4‐丙基苯酚,即稱取0.3410g(2.5mmol)4‐丙基苯酚,同時添加催化劑的量為0.0821g。結果見表5編號2.4‐丙基苯酚在該反應體系下不穩地,有44.4%進一步轉化生成少量正丙苯及環烯、烷烴。表明了該催化劑傾向于保留產物的芳香性,僅有部分的苯環被加氫。
實施例9
本實施例將實施例上述mg1fe2mo‐s催化劑應用于4‐丙基苯酚及甲醇的加氫脫氧反應中。實驗過程類似于實施例4,不同之處在于將反應物替換為4‐丙基苯酚及甲醇,即稱取0.3410g(2.5mmol)4‐丙基苯酚及0.0800g(2.5mmol)甲醇,同時添加催化劑的量為0.0821g。結果見表5編號1.轉化率為32.2%,說明甲醇的加入一定程度上抑制了4‐丙基苯酚的進一步轉化。甲醇對該催化劑提高酚類產物的選擇性具有一定的促進作用。
實施例10
本實施例將上述mg1fe2mo‐s催化劑應用于苯甲醚的加氫脫氧反應中。實驗過程類似于實施例4,不同之處在于將反應物替換為苯甲醚,即稱取0.2704g(2.5mmol)苯甲醚,同時添加催化劑的量為0.0541g。結果見表5編號3.苯甲醚的轉化率為80.9%,產物為苯酚及苯,產率分別為41.9%,33.4%。不同于丁香酚,沒有苯環加氫的產物生成,說明了該反應物比丁香酚更加穩定,并且沒有對位丙基的存在,所得到的芳香烴產物也更加穩定。
實施例11
本實施例將實施例上述mg1fe2mo‐s催化劑應用于愈創木酚的加氫脫氧反應中。實驗過程類似于實施例4,不同之處在于將反應物替換為愈創木酚,即稱取0.3104g(2.5mmol)4‐丙基苯酚,同時添加催化劑的量為0.0621g。結果見表5編號4.愈創木酚的轉化率為61.5%,產物為苯酚及苯,產率分別為33.8%,21.4%。結果類似于苯甲醚,但是相比于苯甲醚,鄰位上酚羥基的存在使得愈創木酚更加難以轉化。
實施例12
本實施例將實施例上述mg1fe2mo‐s催化劑應用于二芐醚的加氫脫氧反應中。實驗過程類似于實施例4,不同之處在于將反應物替換為二芐醚,即稱取0.4957g(2.5mmol)二芐醚,同時添加催化劑的量為0.0991g。結果見表5編號5.催化劑具有很強的c‐o‐c鍵斷鍵能力,能夠比較徹底地打開該二聚體的連接。產物為甲苯,產率為94.2%。
實施例13
本實施例將實施例上述mg1fe2mo‐s催化劑應用于二苯醚的加氫脫氧反應中。實驗過程類似于實施例4,不同之處在于將反應物替換為二苯醚,即稱取0.4255g(2.5mmol)二苯醚,同時添加催化劑的量為0.0851g。結果見表5編號6.該底物相比于二芐醚更加穩定,說明苯環上連接的酚醚c‐o鍵比鏈烴上芐醚c‐o鍵難以斷裂。產物為苯酚,和苯,產率分別為35.6%,55.0%。
實施例14
本實施例將實施例上述mg1fe2mo‐s催化劑應用于芐基苯基醚的加氫脫氧反應中。實驗過程類似于實施例4,不同之處在于將反應物替換為芐基苯基醚,即稱取0.4606g(2.5mmol)基苯基醚,同時添加催化劑的量為0.0921g。結果見表5編號7.結果介于實施例12及13之間,這也是因為芐基苯基醚中既含有酚醚c‐o鍵,也有芐醚c‐o鍵。產物為苯酚,甲苯以及苯,產率分別為37.1%,38.2%,15.8%。
表5其他芳香酚及醚類化合物的催化轉化
- 還沒有人留言評論。精彩留言會獲得點贊!