一種可見光響應型復合光催化劑及其制備方法和應用與流程
本發明涉及光催化技術領域,尤其涉及一種可見光響應型復合光催化劑及其制備方法和應用。
背景技術:
近年來,經濟社會的不斷發展以及工業化進程的不斷深入,導致了大量新興有機污染物廢水的排放,但目前市政污水處理系統難以將其完全去除,從而導致這些新興有機污染物在地表水、地下水、甚至飲用水系統中被檢測出。而這些新興有機污染物,如雙氯酚酸等,即使在痕量濃度下,如若人類及動植物長期暴露其中也會受到嚴重的危害。因此,廢水中新興有機污染物的去除仍是目前環境保護領域的重點和難點。
光催化氧化技術具有反應條件溫和、反應速度快、礦化率高、二次污染少等優勢。美國環保局將其列為最有前景的環保高新技術。tio2基半導體光催化材料是當前國內外研究最為廣泛的光催化劑,但該材料的帶隙較寬(3.2ev),而且只有在紫外光(僅占太陽輻射總量4%)照射下產生光催化活性,這大大限制了它的應用。因此,可高效利用廉價太陽光的可見光響應型光催化劑研制成為目前光催化氧化技術領域的熱點。
氧化鉍(bi2o3)是一種可見光響應型半導體材料,由于其獨特的光學及電學性能,已在氣體傳感器、光伏電池、光學涂層、燃料電池、超級電容等方面有廣泛研究。此外,bi2o3結構中的bi6s軌道孤對電子誘導的內部極化場有助于光生電子-空穴對的分離及載流子的傳遞,從而使bi2o3具有一定的光催化作用。bi2o3主要存在α、β、γ、δ四種晶型結構,其中α-bi2o3常溫下穩定,禁帶寬度約為2.8ev(導帶0.33ev,價帶3.13ev),將其作為可見光響應型光催化劑已有大量研究。然而,雖然β-bi2o3的禁帶寬度為2.4ev,具有比α-bi2o3更強的可見光吸收能力,但是β-bi2o3作為光催化劑的研究仍然存在三方面問題:(1)β-bi2o3在光催化過程中易轉變成α-bi2o3,并與co2反應生成碳酸鹽,導致光催化活性下降;(2)β-bi2o3光催化體系的量子效率低,導致其光催化活性較差;(3)β-bi2o3的導帶低于h+/h2的還原電位,導致光生電子極易與光生空穴復合,從而影響光催化效率。
因此,現有技術還有待于改進和發展。
技術實現要素:
鑒于上述現有技術的不足,本發明的目的在于提供一種可見光響應型復合光催化劑及其制備方法和應用,旨在解決現有β-bi2o3作為光催化劑的研究中存在上述技術缺陷的問題。
本發明的技術方案如下:
一種可見光響應型復合光催化劑,其中,所述可見光響應型復合光催化劑為cubi2o4/β-bi2o3,所述可見光響應型復合光催化劑中cubi2o4為空心微米球,β-bi2o3為不規則納米顆粒,且cubi2o4與β-bi2o3之間緊密接觸。
所述的可見光響應型復合光催化劑,其中,cubi2o4/β-bi2o3中,cubi2o4與β-bi2o3的質量比為1:(0.5-20)。
一種如上任一所述的可見光響應型復合光催化劑的制備方法,其中,包括以下步驟:
(1)cubi2o4的制備:將bi(no3)3·5h2o溶解在濃hno3中,攪拌至完全溶解,再加入cu(no3)2·3h2o,攪拌至混合均勻,而后逐滴滴加0.5-2mol/l的堿性沉淀劑溶液,并將滴加后溶液稀釋,繼續攪拌0.5-2h后,將該溶液轉移至反應釜中,升高溫度至80-150℃,反應18-30h,待反應釜冷卻至室溫后,將反應得到的沉淀物洗滌,并通過離心分離,然后真空干燥,研磨、過篩,即得cubi2o4;
(2)cubi2o4@c的制備:將步驟(1)中所制備的cubi2o4分散于葡萄糖酸溶液中,超聲10-50min后,將混合液移至反應釜中,升高溫度至150-250℃,待反應釜自然冷卻至室溫后,通過離心分離回收反應得到的沉淀物,并將沉淀物洗滌,然后真空干燥,研磨、過篩,即得cubi2o4@c;
(3)cubi2o4/β-bi2o3的制備:將步驟(2)中所制備的cubi2o4@c分散于hno3溶液中,超聲10-50min,得到溶液a;將bi(no3)3·5h2o溶解于hno3溶液中,室溫下劇烈攪拌30-120min,得到溶液b;將堿式碳酸鹽溶解于超純水中,攪拌10-50min,得到溶液c;先將溶液a和溶液b混合后磁力攪拌10-50min,再向其中逐滴滴加溶液c,繼續攪拌10-50min,將反應產生的沉淀物洗滌,并通過離心分離收集沉淀物;將洗滌后的沉淀物放入程序升溫爐,設置程序升溫爐在10-60min內升溫至300-800℃,并在程序升溫爐中反應2-10h,待程序升溫爐自然冷卻至室溫后,收集固體沉淀物,即可得到cubi2o4/β-bi2o3。
所述的可見光響應型復合光催化劑的制備方法,其中,步驟(1)中,所述堿性沉淀劑溶液中的堿性沉淀劑為氫氧化鈉、氫氧化鉀和氨水中的一種或多種。
所述的可見光響應型復合光催化劑的制備方法,其中,步驟(2)中,所述葡萄糖酸溶液中葡萄糖酸的體積為0.04-0.8ml。
所述的可見光響應型復合光催化劑的制備方法,其中,步驟(3)中,所述bi(no3)3·5h2o加入的摩爾量為0.1-3.6mmol。
所述的可見光響應型復合光催化劑的制備方法,其中,步驟(3)中,所述堿式碳酸鹽與bi(no3)3·5h2o的摩爾量之比為(1-10):1。
所述的可見光響應型復合光催化劑的制備方法,其中,步驟(3)中,所述程序升溫爐的溫度升高至300-800℃。
所述的可見光響應型復合光催化劑的制備方法,其中,步驟(3)中,所述程序升溫爐的反應時間為2-10h。
一種可見光響應型復合光催化劑的應用,其中,將如上任一所述的可見光響應型復合光催化劑應用于處理含有非甾體類抗炎藥的廢水。
有益效果:相較純β-bi2o3,本發明所制備的可見光響應型復合光催化劑具有可見光吸收強度更高、光催化性能更好、循環利用率更高等優點。
附圖說明
圖1為本發明實施例1中cubi2o4/β-bi2o3的掃描電子顯微鏡圖。
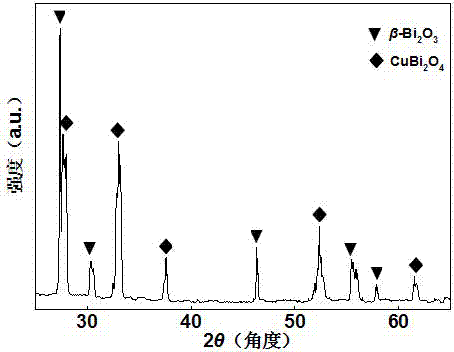
圖2為本發明實施例1中cubi2o4/β-bi2o3的x射線衍射圖譜。
圖3為本發明實施例1中cubi2o4/β-bi2o3的紫外-可見漫反射光譜圖。
圖4為本發明實施例1中cubi2o4/β-bi2o3的光催化性能示意圖。
圖5為本發明實施例9中cubi2o4/β-bi2o3重復利用性能示意圖。
具體實施方式
本發明提供一種可見光響應型復合光催化劑及其制備方法和應用,為使本發明的目的、技術方案及效果更加清楚、明確,以下對本發明進一步詳細說明。應當理解,此處所描述的具體實施例僅僅用以解釋本發明,并不用于限定本發明。
為提高β-bi2o3的光催化活性,將其與其它具有合適能帶結構的半導體復合構筑復合光催化劑是一種有效的技術手段。一方面,可利用兩種半導體不同的能帶結構差異所形成的新的能帶結構特征,改善復合光催化體系中光生電子-空穴的遷移規律,從而減小光生電子-空穴的復合率,提高β-bi2o3的光催化活性;另一方面,還可借助其它半導體的化學穩定性來改善β-bi2o3的結構穩定性。cubi2o4半導體具有可見光響應強、化學穩定性好等優勢,并且其導帶位置較高,其光生電子具有較強的還原能力。此外,相較β-bi2o3,cubi2o4的導帶和價帶勢能均較負,這兩種半導體復合勢必會改變整個反應體系的光催化性能。因此,本發明通過將cubi2o4與β-bi2o3復合構筑復合型光催化劑來提高β-bi2o3的光催化活性和結構穩定性。
具體地,本發明提供一種可見光響應型復合光催化劑,其中,所述可見光響應型復合光催化劑為cubi2o4/β-bi2o3,所述可見光響應型復合光催化劑中cubi2o4為空心微米球,β-bi2o3為不規則納米顆粒,且cubi2o4與β-bi2o3之間緊密接觸。即本發明所述可見光響應型復合光催化劑為cubi2o4/β-bi2o3,所述cubi2o4/β-bi2o3是由空心微米球cubi2o4與不規則納米顆粒β-bi2o3緊密接觸而形成的復合光催化劑。
本發明cubi2o4/β-bi2o3中,cubi2o4與β-bi2o3的質量比為1:(0.5-20)。優選地,cubi2o4與β-bi2o3的質量比為1:(1-15)。更優選地,cubi2o4與β-bi2o3的質量比為1:(1.5-10)。再優選地,cubi2o4與β-bi2o3的質量比為1:(1.8-5)。再進一步優選地,cubi2o4與β-bi2o3的質量比為1:(2-3),例如,cubi2o4與β-bi2o3的質量比為1:2.25。
本發明還提供一種如上任一所述的可見光響應型復合光催化劑的制備方法,其包括以下步驟:
(1)cubi2o4的制備:將bi(no3)3·5h2o溶解在濃hno3中,攪拌至完全溶解,再加入cu(no3)2·3h2o,攪拌至混合均勻,而后逐滴滴加0.5-2mol/l的堿性沉淀劑溶液,并將滴加后溶液稀釋,繼續攪拌0.5-2h后,將該溶液轉移至反應釜中,升高溫度至80-150℃,反應18-30h,待反應釜冷卻至室溫后,將反應得到的沉淀物洗滌,并通過離心分離,然后真空干燥,研磨、過篩,即得cubi2o4。
上述步驟(1)中,所述堿性沉淀劑溶液中的堿性沉淀劑可以為氫氧化鈉、氫氧化鉀和氨水中的一種或幾種。
本發明上述步驟(1)采用水熱法制備形貌結構均勻的cubi2o4。上述步驟(1)具體為,將0.04摩爾份的bi(no3)3·5h2o溶解在2-6ml濃hno3中,優選2-5ml,進一步優選2.5-4ml(如3ml),攪拌使其完全溶解,再加入20ml0.02摩爾份的cu(no3)2·3h2o,攪拌使其混合均勻,而后逐滴滴加0.5-2mol/l的堿性沉淀劑溶液,優選0.8-1.5mol/l的堿性沉淀劑溶液,進一步優選1-1.4mol/l(如1.2mol/l)的堿性沉淀劑溶液,并將滴加后溶液稀釋至50-100ml,優選60-80ml(如70ml),繼續攪拌0.5-2h,優選攪拌0.8-1.5h后,將該溶液轉移至高壓反應釜中,升高溫度至80-150℃,優選90-110℃(如100℃),反應18-30h,優選22-26h(如24h),待反應釜自然冷卻至室溫后,將反應得到的沉淀物洗滌(優選采用超純水反復超聲洗滌),并通過離心分離(轉速優選用5000-7000r/min),然后真空干燥(優選在40-80℃真空干燥箱中干燥6-18h,如12h),研磨、過篩(優選過60-120目篩,如80目篩),即得cubi2o4。
(2)cubi2o4/β-bi2o3的制備:將步驟(1)中所制備的cubi2o4分散于葡萄糖酸溶液中,超聲10-50min后,將混合液移至反應釜中,升高溫度至150-250℃,待反應釜自然冷卻至室溫后,通過離心分離回收反應得到的沉淀物,并將沉淀物洗滌,然后真空干燥,研磨、過篩,即得cubi2o4@c。
步驟(2)中,所述葡萄糖酸溶液中葡萄糖酸的體積為0.04-0.8ml,優選0.1-0.6ml,進一步優選0.2-0.4ml。
本發明上述步驟(2)采用水熱法制備cubi2o4@c。上述步驟(2)具體為,將步驟(1)中所制備的cubi2o4分散于70ml葡萄糖酸溶液中(含葡萄糖酸的體積為0.04-0.8ml,優選0.1-0.6ml,進一步優選0.2-0.4ml,如0.3ml),超聲10-50min,優選20-40min(如30min),將混合液移至100ml高壓反應釜中,升高溫度至150-250℃,優選160-200℃(如180℃),反應2-10h,優選2.5-6h,再進一步優選3-5h(如4h),待反應釜自然冷卻至室溫后,通過轉速為4000-10000r/min,優選5000-7000r/min(如6000r/min)的離心機離心分離回收反應得到的固體沉淀物,并將固體沉淀物洗滌(優選用超純水反復超聲洗滌),然后真空干燥(優選在40-80℃真空干燥箱中干燥6-18h,如在60℃真空干燥箱中干燥12h),研磨、過篩(優選過60-120目篩,如過80目篩),即得cubi2o4@c。
(3)cubi2o4/β-bi2o3的制備:將步驟(2)中所制備的cubi2o4@c分散于hno3溶液中,超聲10-50min,得到溶液a;將bi(no3)3·5h2o溶解于hno3溶液中,室溫下劇烈攪拌30-120min,得到溶液b;將堿式碳酸鹽溶解于超純水中,攪拌10-50min,得到溶液c;先將溶液a和溶液b混合后磁力攪拌10-50min,再向其中逐滴滴加溶液c,繼續攪拌10-50min,將反應產生的沉淀物洗滌,并通過離心分離收集沉淀物;將洗滌后的沉淀物放入程序升溫爐,設置程序升溫爐在10-60min內升溫至300-800℃,并在程序升溫爐中反應2-10h,待程序升溫爐自然冷卻至室溫后,收集固體沉淀物,即可得到cubi2o4/β-bi2o3。
步驟(3)中,所述bi(no3)3·5h2o加入的摩爾量為0.1-3.6mmol,優選0.2-2.5mmol,進一步優選0.25-1.5mmol,再優選0.3-1.0mmol,更優選0.35-0.5mmol。
步驟(3)中,所述堿式碳酸鹽為碳酸鈉、碳酸鉀和碳酸銨中的一種或幾種。
步驟(3)中,所述堿式碳酸鹽與bi(no3)3·5h2o的摩爾量之比為(1-10):1,優選(3-8):1,進一步優選(5-7):1。
步驟(3)中,所述程序升溫爐的溫度升高至300-800℃,優選400-700℃,進一步優選500-650℃。
步驟(3)中,所述程序升溫爐的反應時間為2-10h,優選3-7h,進一步優選4-6h。
本發明上述步驟(3)采用液相合成-煅燒相結合的技術制備cubi2o4/β-bi2o3。上述步驟(3)具體為,將步驟(2)中所制備的cubi2o4@c分散于20mlhno3溶液(其中hno3的濃度為0.5-4mol/l,優選0.6-2mol/l,進一步優選0.8-1.5mol/l,如1.0mol/l)中,超聲10-50min,優選20-40min(如30min),得到溶液a;將0.1-3.6mmol,優選0.2-2.5mmol,進一步優選0.25-1.5mmol,再優選0.3-1.0mmol,更優選0.35-0.5mmol(如0.39mmol)bi(no3)3·5h2o加入到20mlhno3溶液(其中hno3的濃度為0.5-4mol/l,優選0.6-2mol/l,進一步優選0.8-1.5mol/l,如1.0mol/l)中,室溫下劇烈攪拌30-120min,優選40-90min(如60min),得到溶液b;將一定摩爾量的堿式碳酸鹽(堿式碳酸鹽與bi(no3)3·5h2o的摩爾量之比為(1-10):1,優選(3-8):1,進一步優選(5-7):1,如6:1)加入到40ml超純水中,攪拌10-50min,優選20-40min(如30min),得到溶液c。先將溶液a和溶液b混合后磁力攪拌10-50min,優選20-40min(如30min),再向其中逐滴滴加溶液c,此時會產生大量白色沉淀,繼續攪拌10-50min,優選20-40min(如30min),通過轉速為4000-10000r/min,優選5000-7000r/min(如6000r/min)的離心機離心分離回收反應得到的固體沉淀物,并將固體沉淀物洗滌(優選用無水乙醇和超純水反復超聲洗滌)。將洗滌后的沉淀物放入程序升溫爐,設置程序升溫爐在10-60min,優選20-40min(如30min)內升溫至300-800℃,優選400-700℃,進一步優選500-650℃(如600℃),并在程序升溫爐中反應2-10h,優選3-7h,進一步優選4-6h(如5h),待程序升溫爐自然冷卻至室溫后,收集固體沉淀物,即可得到cubi2o4/β-bi2o3。
本發明還提供一種可見光響應型復合光催化劑的應用,其中,將如上任一所述的可見光響應型復合光催化劑應用于處理含有非甾體類抗炎藥(如雙氯芬酸)的廢水。
本發明的可見光響應型復合光催化劑的應用方法是向含有非甾體類抗炎藥的模擬廢水(如雙氯芬酸水溶液)中加入可見光響應型復合光催化劑,即cubi2o4/β-bi2o3,先進行暗吸附反應,待達到平衡后進行可見光光照。在應用過程中按一定時間間隔取樣測定廢水中非甾體類抗炎藥(如雙氯芬酸)的濃度。
優選地,在應用中,cubi2o4/β-bi2o3的用量是:廢水中所含的非甾體類抗炎藥(如雙氯芬酸)與cubi2o4/β-bi2o3的質量之比為1:(10-150),優選1:(50-120),進一步優選1:(70-100),如1:80。
本發明首先利用水熱合成法制備形貌結構均勻、化學性質穩定的可見光響應型半導體cubi2o4和核殼結構cubi2o4@c,在此基礎上,通過液相合成-煅燒相結合的技術在程序升溫的條件下得到cubi2o4/β-bi2o3。相較純β-bi2o3,本發明所制備的可見光響應型復合光催化劑具有可見光吸收強度更高、光催化性能更好、循環利用率更高等優點。
下面通過實施例對本發明進行詳細說明。
實施例1
可見光響應型復合光催化劑的制備:
(1)、首先采用水熱法制備形貌結構均勻的cubi2o4,即將0.04摩爾份的bi(no3)3·5h2o溶解在3ml濃hno3中,攪拌使其完全溶解,再加入20ml0.02摩爾份的cu(no3)2·3h2o,攪拌使其混合均勻,而后逐滴滴加20ml1.2mol/l的naoh,并將滴加后的混合液稀釋至70ml,繼續攪拌1h后,將該混合液轉移至高壓反應釜中,升高溫度至100℃,反應24h,待反應釜自然冷卻至室溫后,將反應得到的沉淀物用超純水反復超聲洗滌,并在6000r/min的轉速下離心分離,然后在60℃真空干燥箱中干燥12h,研磨、過80目篩,即得cubi2o4。
(2)、再進一步采用水熱法制備cubi2o4@c。即準確稱取0.1g步驟(1)中所制備的cubi2o4分散于70ml葡萄糖酸溶液中(含有0.3ml葡萄糖酸),超聲30min后,將混合液移至100ml高壓反應釜中,升高溫度至180℃,反應4h,待反應釜自然冷卻至室溫后,將沉淀物用超純水反復超聲洗滌,并在6000r/min的轉速下離心分離,然后在60℃真空干燥箱中干燥12h,研磨、過80目篩,即得質量比為1:1.4的cubi2o4@c。
(3)、最后采用液相合成-煅燒相結合的技術制備cubi2o4/β-bi2o3。準確稱取0.1g步驟(2)中所制備的cubi2o4@c分散于20mlhno3溶液(1mol/l)中,超聲30min使其充分分散,得到溶液a;將0.39mmolbi(no3)3·5h2o加入到20mlhno3溶液(1mol/l)中,室溫下劇烈攪拌1h使其完全溶解,得到溶液b;將2.34mmolna2co3加入到40ml超純水中,攪拌30min,得到溶液c。先將溶液a和溶液b混合后磁力攪拌30min,使其混合均勻,再向其中逐滴滴加溶液c,此時會產生大量白色沉淀,繼續攪拌30min后,用無水乙醇和去離子水洗滌沉淀物。而后將洗滌后的沉淀物放入程序升溫爐,設置程序升溫爐在30min內升溫至600℃,并在600℃反應5h,待程序升溫爐自然冷卻至室溫后,即可得到質量比為1:2.25的cubi2o4/β-bi2o3。所制得cubi2o4/β-bi2o3的sem(掃描電子顯微鏡圖)、xrd(x射線衍射圖譜)、uv-vis(紫外-可見漫反射光譜)表征結果分別見圖1至圖3。由sem可看到,可見光響應型復合光催化劑中cubi2o4為空心微米球,β-bi2o3為不規則納米顆粒,并經xrd分析證明了可見光響應型復合光催化劑的成分主要是cubi2o4和β-bi2o3,uv-vis圖譜則可以看到,cubi2o4/β-bi2o3在整個可見光譜范圍內均有較高的光吸收強度。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度,該可見光響應型復合光催化劑的光催化性能如圖4。由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為89.02%,遠遠高于純β-bi2o3對雙氯芬酸的去除效率(60.19%)。
實施例2
可見光響應型復合光催化劑的制備與實施例1相同。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.2g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度。由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為80.49%。
實施例3
可見光響應型復合光催化劑的制備與實施例1相同,只是葡萄糖酸溶液中葡萄糖酸的體積為0.6ml。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度。由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為78.62%。
實施例4
可見光響應型復合光催化劑的制備:
(1)、cubi2o4的制備與實施例1相同。
(2)、cubi2o4@c的制備與實施例1相同。
(3)、最后,采用液相合成-煅燒相結合的技術制備cubi2o4/β-bi2o3。準確稱取0.1g步驟(2)中所制備的cubi2o4@c分散于20mlhno3溶液(1mol/l)中,超聲30min使其充分分散,得到溶液a;將1.29mmolbi(no3)3·5h2o加入到20mlhno3溶液(1mol/l)中,室溫下劇烈攪拌1h使其完全溶解,得到溶液b;將7.74mmolna2co3加入到40ml超純水中,攪拌30min,得到溶液c。先將溶液a和溶液b混合后磁力攪拌30min,使其混合均勻,再向其中逐滴滴加溶液c,此時會產生大量白色沉淀,繼續攪拌30min后,用無水乙醇和去離子水洗滌沉淀物。而后將洗滌后的沉淀物放入程序升溫爐,設置程序升溫爐在30min內升溫至600℃,并在600℃反應5h,待程序升溫爐自然冷卻至室溫后,即可得到質量比為1:6的cubi2o4/β-bi2o3。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度。由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為81.36%。
實施例5
可見光響應型復合光催化劑的制備與實施例1相同,只是程序升溫爐的反應溫度為500℃。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度。由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為85.27%。
實施例6
可見光響應型復合光催化劑的制備與實施例3相同,只是程序升溫爐的反應溫度為700℃。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度。由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為83.78%。
實施例7
可見光響應型復合光催化劑的制備與實施例1相同,只是程序升溫爐的反應時間為3h。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度。由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為75.92%。
實施例8
可見光響應型復合光催化劑的制備與實施例1相同。
可見光響應型復合光催化劑應用于去除水中雙氯芬酸的性能測試:在1l20mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度,由測試結果可知,可見光響應型復合光催化劑對雙氯芬酸的去除效率為72.46%。
實施例9:
可見光響應型復合光催化劑的制備與實施例1相同。
可見光響應型復合光催化劑重復應用于去除水中有機物的性能測試:在1l5mg/l雙氯芬酸溶液中,投加0.4g上述可見光響應型復合光催化劑,先進行暗吸附反應30min達到吸附平衡后,再在300w氙燈照射條件下光催化反應3h,實驗結束后通過離心使固液分離,并測定上清液中雙氯芬酸殘余濃度。回收的可見光響應型復合光催化劑經超純水洗滌數次、在60℃真空干燥箱中干燥后,研磨,過80目篩,再次應用于雙氯芬酸廢水處理,處理過程同上,其重復利用效率如圖5所示,由測試結果可知,可見光響應型復合光催化劑重復利用第七次時,其對雙氯芬酸的降解效率為78.43%。
綜上所述,本發明提供的一種可見光響應型復合光催化劑及其制備方法和應用,本發明首先利用水熱合成法制備形貌結構均勻、化學性質穩定的可見光響應型半導體cubi2o4和核殼結構cubi2o4@c,在此基礎上,通過液相合成-煅燒相結合的技術在程序升溫的條件下得到cubi2o4/β-bi2o3。本發明所制備的可見光響應型復合光催化劑在整個可見光譜范圍內均有較強吸收,且相較純β-bi2o3具有更好的光催化性能和更高的循環利用率。將0.4g的cubi2o4/β-bi2o3(1:2.25,wt%)用于處理雙氯芬酸難降解有機廢水,可見光照射3h,對1l5mg/l雙氯芬酸溶液的去除率為89.02%,重復利用第七次,對雙氯芬酸溶液去除率為78.43%。
應當理解的是,本發明的應用不限于上述的舉例,對本領域普通技術人員來說,可以根據上述說明加以改進或變換,所有這些改進和變換都應屬于本發明所附權利要求的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種普魯士藍/三氧化鎢復合光催化劑及其制備方法與應用與制造工藝
- 一種具有可見光響應的多功能光電催化膜的制備方法與制造工藝
- 一種以魚類鱗片為模板制備磷酸銀納米顆粒薄膜的方法與制造工藝
- 一種氮摻雜偏銦酸鋰光催化材料的制備方法及其制品的制作方法
- 一種Fe<sub>3</sub>O<sub>4</sub>?C@Bi<sub>2</sub>O<sub>3</sub>?BiOI光催化劑及其制備方法和應用
- 可見光響應的光催化劑Li<sub>3</sub>FeSiO<sub>5</sub>的制作方法
- 一種用于處理廢氣的多孔材料負載氧化銀的制備方法
- 可見光響應的光催化劑LiInSn<sub>3</sub>O<sub>8</sub>及其制備方法
- 可見光響應的光催化劑Li<sub>2</sub>SrMgGeO<sub>5</sub>及其制備方法
- 可見光響應的光催化劑Li<sub>2</sub>CaSnO<sub>4</sub>及其制備方法