新型變色纖維AMF?PAR的制備及應用的制作方法
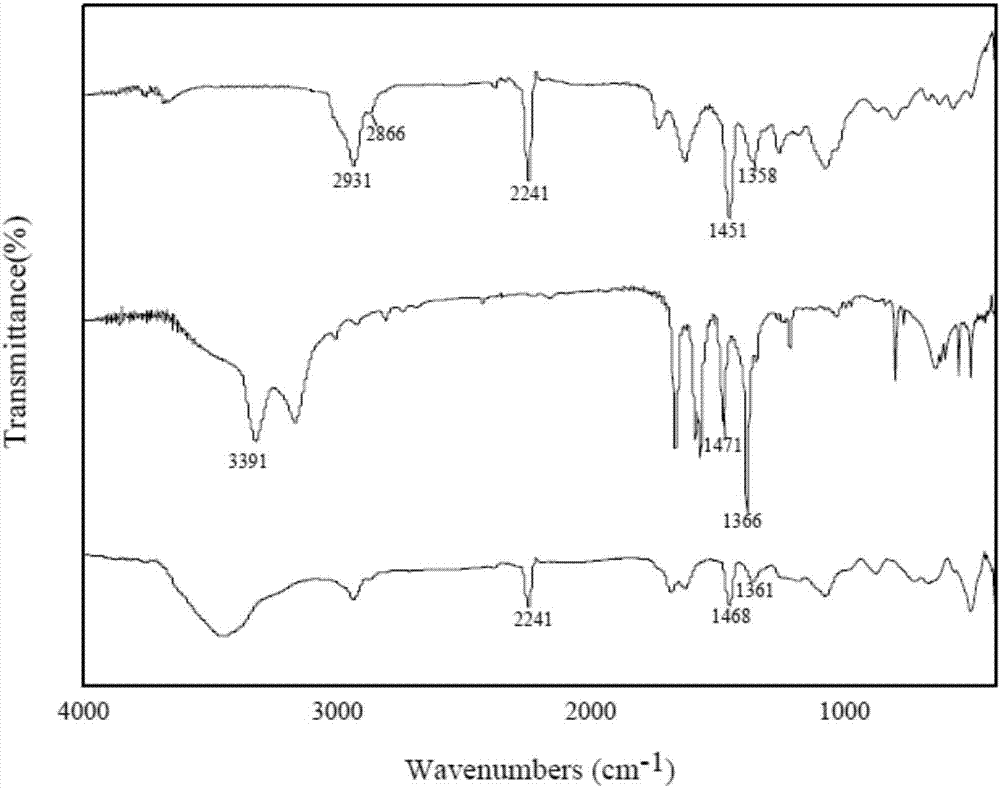
本發明涉及螯合纖維合成領域,尤其涉及一種螯合纖維amf的合成方法及變色纖維amf-par的合成方法及應用。
背景技術:
:螯合纖維是一類以纖維狀聚合物為載體,連接特殊功能基從而與特定物質螯合以實現分離的功能高分子,是繼離子交換樹脂、離子交換纖維發展起來的一種新型高性能吸附材料。近年來,螯合纖維在金屬離子的提取、分離、分析等方面的研究成為分離科學領域的研究熱點。隨著現代分析技術和分析儀器的發展(如icp-ms、icp-aes及aas等),元素的檢測限已經大幅度降低,例如,使用icp-ms可以檢測出ppm級的金屬離子,但痕量和超痕量重金屬元素分析過程中,由于大量共存元素的干擾,直接測定常常很困難,一些分析儀器擁有較高的選擇性及靈敏度,但高昂的檢測成本限制了其在國內中小型食品企業生產檢測中的普及。螯合纖維素作為一種物美價廉,既不產生污染又能有效分離富集重金屬離子的吸附材料,在分析檢測,資源回收等方面有著重要的研究價值和廣泛的應用前景。汞、銅是環境中主要的重金屬污染物。存在于大氣、水體、土壤中的汞、銅可以導致食品污染,對人類的健康和生存都構成巨大威脅。海產品是重金屬污染最為嚴重的食品之一,因為海產品可以通過生物富集作用吸收海水中大量有毒元素。長期食用含汞超標的海產品會損害人體的中樞神經系統,這是因為汞通過食物通道進入人體后與蛋白酶的巰基結合,抑制了酶的活性,從而阻礙細胞的正常代謝,嚴重的還會損傷肝腎功能。食用含銅較多的海產品也會對人肝腎造成傷害,當人體內殘存了大量的銅離子,極易對身體臟器造成巨大負擔,新陳代謝紊亂,肝硬化,肝腹水甚至更為嚴重。目前,海產品中重金屬常規檢測方法有:沉淀分析法、絡合滴定法、分光光度法、原子吸收光譜法、發射光譜分析法等,但這些方法普遍都要求昂貴的儀器,檢測費用高等缺點,同時重金屬檢測是一項十分復雜的工作,為了滿足大量樣品的快速檢測要求,尋找一種具有攜帶方便,成本低廉同時又具備一定靈敏度和準確性的快速檢測方法就變得尤為重要。技術實現要素:本發明的目的在于解決現有技術中存在的上述技術問題,提供一種腈綸螯合纖維amf及其合成方法,以腈綸纖維為母體,與配體(2-氨基-4-甲基嘧啶)進行合成反應,能夠獲得具有較高功能基轉化率的腈綸螯合纖維amf,該腈綸螯合纖維amf對重金屬具有良好的選擇性吸附性能,其中對銅離子和汞離子具有較佳的選擇吸附性。同時提供一種新型腈綸顯色纖維amf-par的合成及其應用,能夠實現對海產品中汞及銅的快速檢測。為了解決上述技術問題,本發明提供以下技術方案:本發明的腈綸螯合纖維amf的合成方法,包括以下步驟:(1)以腈綸纖維(pan)為母體,將腈綸纖維置于反應溶劑水中浸泡溶脹6h;(2)在步驟(1)的所得物中加入配體,在氮氣保護條件下,于30℃~90℃攪拌至反應結束,其中,所述配體為2-氨基-4-甲基嘧啶,母體與配體的摩爾比為1:3~1:6;(3)將步驟(2)的所得物用反應溶劑水洗滌至無色,經沖洗后干燥恒重,得到腈綸螯合纖維amf。作為本發明的腈綸螯合纖維amf的合成方法的改進,所述步驟(1)中,腈綸纖維與水的用量比為0.10-0.20g腈綸纖維/30ml水。作為本發明的腈綸螯合纖維amf的合成方法的改進,所述腈綸纖維與所述2-氨基-4-甲基嘧啶的摩爾比優選為1:4。作為本發明的腈綸螯合纖維amf的合成方法的改進,所述步驟(3)中的沖洗為:經無水乙醇、丙酮、乙醚依次洗滌。作為本發明的腈綸螯合纖維amf的合成方法的改進,所述步驟(2)中的反應溫度優選為90℃。本發明所得的合成纖維即為新型的腈綸螯合纖維amf,根據產物含n量,合成纖維的功能基含量和功能基轉化率可以通過公式(1)和(2)計算:也可以根據含s量,合成纖維的功能基含量及功能基轉化率也可以通過公式(3)和(4)計算:其中,f0為腈綸纖維功能基含量(cnmmol/g),fc為合成纖維的功能基含量(mmol/g),x為功能基轉化率,sc為合成纖維含硫量(%),n0為腈綸纖維含氮量(%),nc為合成纖維含氮量(%),ml為配體的摩爾質量(mol/g),ns為配體分子中含硫原子個數,nn為配體分子中含氮原子個數。本發明的采用腈綸螯合纖維amf制備par顯色纖維的制備方法,將腈綸螯合纖維amf和par置入甲醛水溶液中,在氮氣保護條件下攪拌加熱回流2-6h,于70℃攪拌反應結束,用溫水沖洗后,干燥恒重,得到顯色纖維amf-par;作為本發明的顯色纖維amf-par的合成方法的改進,所述amf、par、水及甲醛的用量比優選為:0.5gamf/0.3gpar/35ml水/5ml甲醛。本發明的合成方法制得的顯色纖維amf-par在海產品中重金屬快速檢測中的應用。所述重金屬為hg2+或cu2+。本發明的腈綸顯色纖維amf-par在海產品中hg(ii)快速檢測中的應用,包括以下步驟:(1)待測樣品前處理:將待測樣品經搗碎、消解、定容后得到待測溶液;所述待測樣品為海產品;(2)將顯色纖維amf-par浸沒在待測溶液內,2s后取出,甩干,顯色完全后,根據顯色情況判斷待測樣品中的重金屬濃度。步驟(2)中,還包括:將顯色纖維amf-par經掩蔽劑處理后,浸沒在待測溶液內;若待檢測的重金屬含hg2+,所述掩蔽劑為edta二鈉鹽。所述重金屬離子cu2+濃度達到500ppm時,掩蔽效果并不明顯,hg2+濃度大于100ppm經過掩蔽加工的amf-par纖維對cu2+的掩蔽效果已經明顯減弱。采用本發明的腈綸螯合纖維amf的合成方法及顯色纖維amf-par,具有以下優點:(1)所得的腈綸螯合纖維amf及腈綸顯色纖維amf-par具有原料來源廣泛,價格低廉易制備等特點;(2)本發明合成的腈綸螯合纖維amf具有重金屬離子吸附能力,其中,對銅離子、汞離子有較強的選擇吸附性,吸附量大,吸附速度快;(3)本發明合成的腈綸螯合纖維amf的洗脫效率高;(4)本發明提供的合成方法操作簡單,產率高;(5)通過本發明合成的腈綸顯色纖維amf-par具有足夠的拉伸斷裂強度以滿足實際應用需求,可以適用于不同環境下的檢測,能夠制成不同形態的顯色材料;(6)本發明合成的腈綸顯色纖維amf-par對銅離子和汞離子具有明顯的顯色選擇性,能夠用于檢測海產品中含銅或含汞量;(7)本發明提供的重金屬檢測應用,有效降低了檢測成本,簡化了檢測步驟,可以應用于食品中微量汞的檢測分析。附圖說明圖1所示為實施例1制備的amf的紅外光譜圖;圖2所示為不同反應溫度對amf功能基轉化率的影響;圖3所示為不同反應摩爾比對amf功能基轉化率的影響;圖4所示為不同ph下amf對六種重金屬離子的吸附量;圖5所示為amf在不同溫度下對hg(ⅱ)金屬離子的吸附量隨時間的變化曲線;圖6所示為amf在不同溫度下對cu(ⅱ)金屬離子的吸附量隨時間的變化曲線;圖7所示為顯色纖維amf-par對不同金屬的顯色效果示意圖;圖8所示為顯色纖維amf-par在不同ph下對hg(ⅱ)的顯色效果示意圖;圖9所示為amf-par在不同濃度cu(ⅱ)干擾下對10ppmhg(ⅱ)的顯色效果示意圖;圖10所示為amf-par在不同濃度cu(ⅱ)干擾下對50ppmhg(ⅱ)的顯色效果示意圖;圖11所示為amf-par在不同濃度cu(ⅱ)干擾下對100ppmhg(ⅱ)的顯色效果示意圖。具體實施方式下面結合附圖和具體實施方式對本發明作進一步詳細的說明。實施例1(1)準確稱取0.15g母體腈綸纖維(pan)置于100ml三頸瓶中,再加入30ml反應溶劑水,浸泡溶脹6h;(2)向三頸瓶中加入配體2-氨基-4-甲基嘧啶,并接通氮氣,以150rpm的攪拌速度在常溫下攪拌1-2h,待瓶內空氣排凈后(即在氮氣保護條件下),于90℃攪拌至反應結束,其中,腈綸纖維(母體)與2-氨基-4-甲基嘧啶(配體)的摩爾比為1:4;(3)待步驟(2)中反應結束后,將步驟(2)的所得物用水洗滌至無色,再依次用無水乙醇、丙酮、乙醚洗滌數次,于50℃條件下真空干燥至恒重,得到腈綸螯合纖維amf。圖1所示為實施例1制備的amf的紅外光譜圖;2241m-1峰是腈綸纖維中氰基cn鍵的伸縮振動吸收峰,2931m-1峰為腈綸分子骨架中-ch2的不對稱伸縮振動峰,而2866cm-1峰則為對稱伸縮振動峰,1451m-1、1358cm-1處為c-h的面內彎曲振動吸收峰,由圖分析可知,經過am改性,氰基因反應消耗而減少,腈綸纖維母體中2241cm-1附近c≡n鍵吸收峰明顯減弱;對比pan紅外圖譜可以發現,1468cm-1,1361cm-1處的吸收峰表明了amf中雜環的存在。即雜環中-ch2單鍵振動峰,運用紅外光譜技術方法,通過對反應前后腈綸纖維的對比分析,得出腈綸螯合纖維amf合成反應的反應路徑及amf結構如下所示:對比例1-1將實施例1中的反應溶劑改成甲苯、正丁醇,其余同實施例1。具體而言,本實施例中,以腈綸纖維為母體,由于極性很大的氰基相互作用,腈綸纖維中的丙烯腈具有不規則的空間立體結構,從而導致腈綸纖維分子間的作用力很大。有機反應溶劑對許多高分子材料有溶脹作用,有利于化學改性的進行。因此,綜合考慮腈綸纖維在反應溶劑中的溶脹性、溶解性以及溶劑的沸點、極性,以水、甲苯、正丁醇作為反應溶劑進行比較,結果見表1。表1三種不同溶劑下amf的元素分析根據表1,綜合比較amf的含氮量和含硫量,2-氨基-4-甲基嘧啶在甲苯和水中都有較高的功能基轉化率,但無論從成本和安全性上看,廉價清潔的水是更好的溶劑選擇。對比例1-2將實施例1步驟(2)中的反應溫度90℃改為30℃、50℃、70℃的條件下進行攪拌,其余完全同實施例1的步驟相同,從而探討反應溫度對amf功能基轉化率的影響。具體而言,高分子材料的運動力有不同形式,絕大多數有機高聚物材料都有四種物理狀態:玻璃態、粘彈態、高彈態、粘流態,高彈態和玻璃態之間的轉變稱為玻璃化轉變。在玻璃化轉變溫度以下,高聚物處于玻璃態,分子鏈和鏈段都不能動,不利于化學改性的進行。腈綸纖維的玻璃化轉化溫度為80-100℃。所以,當反應溫度大于腈綸的玻璃化轉化溫度,反應才能順利進行。相反反應溫度過高,纖維結構容易遭到破壞。溶劑水、甲苯、正丁醇的沸點分別為100℃、110.6℃、117.7℃,配體的沸點為186℃。綜合考慮溶劑以及配體的熔沸點,amf在水中反應溫度范圍選擇在30℃~90℃,如圖2所示,在反應溫度范圍內,功能基轉化率均隨溫度升高而升高,所以amf的最佳合成溫度為90℃。對比例1-3將實施例1步驟(2)中,母體與配體的反應摩爾比改為1:2,1:3,1:5,1:6,其余同實施例1,從而探討反應摩爾比對amf功能基轉化率的影響,結果如圖3所示。根據圖3可知,隨著摩爾比增加,amf功能基轉化率先升高再降低,于1:4時,功能基轉化率達到最大值。因此,確定最佳反應摩爾比為1:4。綜上所述,amf的最佳合成條件如表2所示。表2amf最佳合成條件此外,采用質構儀對腈綸纖維及改性后的腈綸螯合纖維amf的拉伸斷裂強度進行初步的測試。從測試結果得出:未經任何改性的聚丙烯腈纖維pan單絲平均斷裂拉力(g)為8.32,經過改性后的腈綸螯合纖維amf單絲平均斷裂拉力(g)為5.74,其斷裂強度仍然能達到腈綸纖維原始拉伸斷裂強度的69%,這充分說明了改性后的腈綸螯合纖維有足夠的拉伸斷裂強度去滿足實際應用的需求。實驗1-1稱取15.0mg腈綸螯合纖維amf多份,放入100ml碘量瓶中,移入25ml不同ph的hac-naac緩沖液,放置6h,加入5ml金屬離子標液,25℃,100rmp,振蕩吸附平衡。取樣,測定溶液中殘留金屬離子濃度。用下式計算吸附量(q)、吸附率(e):式中q為腈綸螯合纖維吸附容量(mg/g);c0和ce分別為金屬離子的初始濃度(mg/ml)和平衡濃度(mg/ml);w為腈綸螯合纖維質量(g);v為金屬離子溶液體積(ml)。于本實驗中,ph范圍為2.5~6.5,金屬離子標液分別為重金屬離子zn(ⅱ)、pb(ⅱ)、cd(ⅱ)、ni(ⅱ)、cu(ⅱ)、hg(ⅱ)的標液。如圖4所示,腈綸螯合纖維amf對hg(ⅱ)、cu(ⅱ)的吸附量相比其他四種重金屬的吸附量大很多,具有良好的選擇吸附性。另外,如圖4所示,溶劑ph值對腈綸螯合纖維amf吸附hg(ⅱ)的影響較大,最佳吸附ph為6.5。溶液的ph影響重金屬在水中的存在狀態(分子、離子、絡合物)及溶解度,同時影響著纖維上功能基質子化的程度。酸性較強的環境中,纖維上功能基質子化程度加強,不利于重金屬離子吸附;而在堿性較強的條件下,重金屬離子的溶解度減小,易發生沉淀。所以強酸強堿環境均不利于螯合纖維對重金屬離子的吸附。腈綸螯合纖維amf對cu(ⅱ)的吸附受ph影響較小,而這一性質在實際應用中有著積極的作用,可以避免由于吸附條件的苛刻限制了腈綸螯合纖維的使用范圍。實驗1-2稱取15.0mg腈綸螯合纖維amf多份,置于100ml碘量瓶中,移入25ml最佳吸附ph的hac-naac緩沖液,放置6h,加入5ml金屬離子標液,分別在15℃、25℃和35℃下,100rmp,振蕩吸附。定時取樣,測定溶液中金屬離子濃度,直至溶液中金屬離子濃度不變,達到吸附平衡。于本實驗中,金屬離子標液為重金屬離子cu(ⅱ)、hg(ⅱ)的標液。如圖5及圖6所示,吸附量(q)隨著時間的增長而變大,并在某一時間點達到平衡。在前15min內,腈綸螯合纖維上的吸附位點較多,且溶液中的金屬離子的濃度較高,較大的傳質動力使得吸附速率較快,吸附量增加迅速。隨著腈綸螯合纖維上可結合的活性位點吸附飽和,吸附空間阻礙變大,溶液中金屬濃度的降低,吸附速率逐漸降低,最后達到吸附平衡。腈綸螯合纖維amf對hg(ⅱ)的吸附平衡時間為20min;對cu(ⅱ)的吸附平衡時間為22min。相比離子交換樹脂等其他傳統吸附材料,吸附平衡的時間被大大縮短,通常使用的離子交換樹脂需要數個小時才能達到吸附飽和。這是因為腈綸螯合纖維的單絲直徑只有20-30微米,金屬離子在纖維中的傳質距離短;同時擁有更大的比表面積,所以腈綸螯合纖維具有較好的動力學性能。從圖中還可以看出溫度對吸附速率和吸附量也存在影響。隨著溫度升高,纖維的吸附速率和吸附量皆依次升高,說明在實驗溫度范圍之內,溫度的升高有利于吸附的進行。實驗1-3:靜態解吸實驗稱取15.0mg腈綸螯合纖維amf多份,放入100ml碘量瓶中,移入25ml最佳吸附ph的hac-naac緩沖液,放置6h,加入5ml金屬離子標液,25℃,100rmp,振蕩吸附平衡。取樣,測定溶液中殘留金屬離子濃度。將吸附平衡后的纖維過濾,用去離子水洗滌多次,晾干后放入新的100ml碘量瓶中,加入30ml解吸劑,25℃,100rmp,振蕩至解吸平衡測定溶液中重金屬離子濃度(ce')。解吸率(e'):式中c0和ce分別為吸附階段金屬離子的初始濃度(mg/ml)和平衡濃度(mg/ml);ce'為解吸平衡后錐形瓶中金屬離子的濃度。于本實驗中,金屬離子標液為重金屬離子cu(ⅱ)、hg(ⅱ)的標液。解析劑為不同濃度的hcl和hno3。實驗結果如表3及表4所示。表3兩種不同解吸劑對amf吸附hg(ⅱ)的解吸率表4兩種不同解吸劑對amf吸附cu(ⅱ)的解吸率由表3和表4可知,解吸劑的種類和濃度對解析效果均有較大的影響。在對hg(ⅱ)的解吸中,3.0mol/l的hno3可以完全解吸amf,在對cu(ⅱ)的解吸實驗中,3.0mol/l的hno3同樣可以使amf的解吸率達到100%。實施例2將0.5g腈綸螯合纖維amf、0.3gpar、35ml水和5ml甲醛水溶液投入100ml三頸瓶中,在氮氣保護下攪拌加熱回流4h,于70℃攪拌反應結束,取出纖維,用溫水反復沖洗纖維至中性,放入烘箱干燥(50℃)2h,得到顯色纖維amf-par。于室溫下,將顯色纖維amf-par分別置于多種重金屬離子溶液中,觀察顯色效果。其中,重金屬離子溶液包括pb2+、zn2+、cu2+、ni2+、hg2+、cd2+。如圖7所示,顯色纖維amf-par對10-2mol/l的pb2+、zn2+、cu2+、ni2+、hg2+、cd2+六種重金屬進行顯色,我們可以清晰得看出,在ph值為6條件下,顯色纖維amf-par對cu2+、hg2+都有很明顯的顯色作用,顏色從橙黃變回深紫紅色,而對其他四種重金屬不顯色,說明了amf-par具有較好的顯色選擇性。同時在試驗中發現,在相同環境條件下,amf-par對等濃度hg2+的顯色速度明顯比cu2+快。本發明中,變色纖維amf-par的合成原理化學式如下:另外,圖8所示為顯色纖維amf-par在不同ph下對hg(ⅱ)的顯色效果示意圖。如圖8所示,ph對顯色纖維amf-par遇到金屬時顯色具有較大的影響,在ph較低時,顯色纖維amf-par不顯色或顯色不完全,當ph增大到6時,顯色纖維可以得到充分顯色。這是因為,在ph值降低時,纖維上的氨基和par分子上的吡啶基團會因為質子化而降低了對溶液中金屬離子的絡合能力,只有有效的發生絡合作用才能使纖維變色。當ph大于6時,溶液中金屬離子易產生沉淀,使顯色過程不穩定,影響顯色實驗結果。此外,通過纖維拉伸斷裂強度測試可得到如表5所示結果。表5pan、amf和amf-par拉伸斷裂強度測試panamfamf-par單絲平均斷裂拉力(g)8.325.745.09改性后強度占pan強度百分比6961.2由表5可知,經過化學改性和接顯色劑之后的腈綸纖維強度并沒有發生太大減弱,可以適用于不同環境下的檢測,同時可以制成不同形態的顯色材料。對比例2-1將實施例2中par改為8-羥基喹啉、鉻黑t,其余同實施例2相同;所得顯色纖維分別為顯色纖維i、顯色纖維ii,其與本發明的顯色纖維amf-par的對比結果如表6所示。表6不同顯色纖維對重金屬離子顯色顯色纖維顯色結果顯色纖維amf-par對hg2+、cu2+兩種金屬離子有顯色顯色纖維i:8-羥基喹啉顯色纖維無響應顯色纖維ii:鉻黑t顯色纖維無響應實驗2-1由實施例2顯示,腈綸顯色纖維amf-par只對hg2+、cu2+兩種金屬離子有顯色作用。然而,共存金屬離子是影響顯色反應結果準確性的重要因素。故本實驗考察cu2+對hg2+在顯色纖維上的顯色干擾影響,從而確定顯色纖維法測定水樣中共存cu2+的最大濃度。配制不同濃度的cu2+標準溶液和hg2+標準溶液,將掩蔽劑edta二鈉鹽配制成掩蔽劑溶液。向某一濃度的hg2+標準溶液中加入不同濃度的cu2+標準溶液,將顯色纖維amf-par分別浸沒在掩蔽劑溶液中1min左右,取出干燥。再把干燥好的顯色纖維amf-par放入上述不同的溶液中,觀察比色管中纖維顏色變化。圖9所示為amf-par在不同濃度cu(ⅱ)干擾下對10ppmhg(ⅱ)的顯色效果示意圖。如圖9所示,第1管中未加cu2+且amf-par顯色纖維未浸過掩蔽劑,第2、3、4管在第1管的基礎上加入了不同濃度的cu2+,后三管加了不同濃度的cu2+同時amf-par顯色纖維經過了掩蔽劑的處理。從圖9中可以看出,第1管中amf-par顯色纖維遇到hg2+后正常顯色,未加掩蔽劑的三管amf-par纖維顏色已經從橙黃色變成了紫紅色,即cu2+嚴重干擾了amf-par顯色纖維對目標離子hg2+的顯色,而經過掩蔽劑溶液處理過的amf-par顯色纖維與第1管中顯色結果基本一致,排除了cu2+對顯色的干擾。圖10所示為amf-par在不同濃度cu(ⅱ)干擾下對50ppmhg(ⅱ)的顯色效果示意圖。如圖10所示,增加了hg2+的濃度,實驗結果與圖9類似,經過掩蔽劑加工的amf-par顯色纖維可以很好得掩蔽cu2+對實驗結果的干擾。但當cu2+濃度達到500ppm時,掩蔽效果并不明顯。圖11所示為amf-par在不同濃度cu(ⅱ)干擾下對100ppmhg(ⅱ)的顯色效果示意圖。如圖11所示,當hg2+的濃度增加到100ppm,7支比色管中的amf-par顯色纖維顏色區別明顯減小。說明溶液中的hg2+濃度大于100ppm,經過掩蔽加工的amf-par纖維對cu2+的掩蔽效果已經明顯減弱。實驗2-2amf-par纖維測試件上有par,如果par在很短的時間內變質而導致測試件無法正常使用,這樣保存時間短的纖維測試件不能作為有效的測試工具。所以將制備好的amf-par測試件分別放在避光和自然光下,一部分置于室溫保存,一部分置于低溫(5-10℃)保存,每個半個月取出,與標準比色板比較,測定其顯色性能是否有衰退。實驗發現,隨著時間的增加,在室溫下自然光保存的腈綸顯色纖維amf-par本身顏色不斷變深,2個月后,測定10mg/lhg(ⅱ)標準溶液,顏色色階與標準比色板不符,所以自然光下室溫存儲時間為2個月。同樣在室溫下,避光保存的腈綸顯色纖維amf-par可以穩定存儲6個月;而在低溫條件下(5-10℃),存儲時間可以延長至1年。實驗2-3配制一系列不同濃度的hg(ⅱ)標準溶液0mg/l、0.1mg/l、0.2mg/l、0.3mg/l、0.4mg/l、0.5mg/l、1mg/l、2mg/l、3mg/l、4mg/l、5mg/l、10mg/l、20mg/l、50mg/l、100mg/l,將制備好的腈綸顯色纖維amf-par完全浸沒在上述不同的標準溶液中,2s后取出甩干。用數碼相機拍攝輸入電腦,用photoshopcs4吸管工具將纖維顏色值轉化為rgb值,并記錄如表7所示。將顯色完畢的amf測試件通過數碼相機拍攝并把圖像導入電腦,用photoshopcs4軟件中吸管工具將測試件的顏色轉化為rgb模式(r代表red(紅色)、g代表green(綠色)、b代表blue(藍色))信息,數值在0-255之間。把顏色數值化,為標準比色板的制作提高了準確性。表7amf-par標準比色板由表7可以看出,比色板中整體顏色從淺到深,而且不同取樣點之間顏色均勻,數據相差不會超過10。實施例3(1)制備腈綸螯合纖維amf:具體過程如實施例1中amf的制備過程。(2)制備顯色纖維amf-par:具體過程如實施例2中amf-par的制備過程。(3)待測樣品前處理:稱取搗碎均質后的金槍魚樣品0.5g置于聚四氟乙烯管中,加入1ml30%h2o2,10mlhno3,然后放入消解罐中,置于消解儀中,設定消解條件:800w,15min;1000w,25min;0w,15min。消化完取出聚四氟乙烯管放在電熱板上,120℃加熱2h,趕酸至消解液剩余約1ml,移入容量瓶,用去離子水定容至25ml,待測。(4)將經過掩蔽制備后的amf-par腈綸纖維完全浸沒在比色管中,2s后取出,甩干,顯色完全后將顏色轉化為rgb值,與標準比色卡對比得到溶液中hg(ⅱ)濃度。對比例3-1配制不同濃度的hg2+標準溶液各10ml,取10ml金槍魚、馬頭魚消解后定容的樣品溶液,用icp-aes測定樣品溶液中hg(ⅱ)濃度。表8amf-par比色檢測法與icp-aes檢測法比較參照國標gb2762-2012可知,隨機抽樣的兩種海產品:金槍魚中可食部分的汞含量未超標;馬頭魚可食部分中的汞含量已經超標。顯色纖維amf-par測得海產品中hg(ⅱ)濃度相差不大,且與常規檢測儀器icp-aes檢測結果基本一致,相近的添加回收率也說明了用改性腈綸螯合顯色纖維amf-par檢測海產品中汞含量是切實可行的,表明該方法具有準確度高,分析結果可靠等特點。同時相比常規的重金屬檢測方法,有效得率降低了檢測成本,簡化了檢測步驟,可以應用于食品中其他樣品中微量汞的檢測分析。上述實施例不以任何方式限制本發明,凡是采用等同替換或等效變換的方式獲得的技術方案均落在本發明的保護范圍內。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1