一種表面功能化的微米級多孔順磁性球形樹脂顆粒的制備方法與流程
本發明涉及固體材料技術領域,具體涉及一種不同表面功能化的微米級多孔順磁性球形樹脂顆粒的制備方法。
背景技術:
磁性材料,特別是磁性納米材料,因其較高的比表面積和順磁特性使其出在快速、簡便的進行分離富集物質時展現出巨大優勢。但納米材料在使用時也具有明顯的不足和缺陷,包括制備方法不夠簡便、穩定,顆粒易團聚,溶液中的殘留易堵塞過濾裝置等問題。而微米級的功能化的樹脂多孔顆粒具有高強度、高比表面、耐酸堿等優良特性,在醫藥、生物和化學化工等領域被廣泛用做分離、分析和富集材料。但相對磁性材料,單純的樹脂顆粒無法依靠磁場進行快速的分離和轉移。制備具有順磁特性的微米級功能化的多孔樹脂顆粒的材料則能有效的結合前述兩種材料的優勢,使其在分離富集及其與自動化儀器結合時帶來更大的優勢。
技術實現要素:
本發明提供了一種具有順磁性能的聚合物微球的制備方法;所述方法包括如下步驟:
(1)通過硝化試劑對聚合物微球表面進行硝化處理;
(2)將步驟(1)得到的微球與亞鐵鹽或亞鐵鹽與其他金屬的鹽的組合、和堿進行反應;
優選的,所述的聚合物微球為乙烯基單體聚合得到,可以為一種乙烯基單體的均聚物,也可以兩種以上的乙烯基單體的共聚物;所述的乙烯基單體優選自:苯乙烯、馬來酸酐、丙烯酸酯、乙烯基酯、及其衍生物,更優選自:苯乙烯、丙烯酸甲酯、甲基丙烯酸甲酯、丙烯酸乙酯、甲基丙烯酸乙酯、丙烯酸丁酯、甲基丙烯酸丁酯、乙酸乙烯酯中的一種或多種;
在本發明的一個優選實施例中,所述的乙烯基單體為苯乙烯,所述的聚合物微球為苯乙烯的均聚物;
上述步驟(1)中所用的聚合物微球可采用現有技術中公開的方法制備得到,也可以采用商購;
優選的,所述的聚合物微球為多孔交聯的;優選的,聚合物微球的交聯度為40-80%;更優選為60-80%;
優選的,所述的聚合物微球為球形;
優選的,所述的微球的粒徑為100-400目,更優選為200-400目;
優選的,所述的微球的比表面積100-600m2/g,更優選為300-500m2/g;
優選的,所述的微球的孔徑為3-50nm,更優選為3-10nm;
優選的,所述的硝化試劑優選自:濃硝酸(65-70wt%)、濃硝酸(65-70wt%)-濃硫酸(>98wt%)的混合液、濃硝酸(65-70wt%)-冰醋酸的混合液;
優選的,所述的濃硝酸-濃硫酸的混合液為濃硝酸與濃硫酸以體積比1:1的混合液;
優選的,所述的濃硝酸-冰醋酸的混合液為濃硝酸與冰醋酸以體積比1:1的混合液;
優選的,所述的亞鐵鹽為氯化亞鐵或硫酸亞鐵,如四水合氯化亞鐵或七水合硫酸亞鐵;
優選的,所述的其他金屬為鎳(Ni)、鈷(Co)、錳(Mn)。
在本發明的一個實施例中,步驟(2)中將步驟(1)得到的微球與亞鐵鹽、堿反應,形成磁性粒子Fe3O4;
優選的,所述的堿為氨水;在本發明一個優選實施例中,所述氨水為飽和氨水;
優選的,所述的聚合物微球與硝化試劑的質量體積比為0.5-10g/ml;更優選為1-5g/ml;
優選的,所述的硝化時間為1-24h,更優選為1-10h;
優選的,所述的硝化溫度為室溫;
優選的,步驟(2)中,步驟(1)得到的微球與亞鐵鹽的質量比例為1:0.6-5;更優選為1:0.8-1.5;
優選的,步驟(2)中,步驟(1)得到的微球與堿的質量體積比為0.1-10g/ml,更優選為0.1-1.0g/ml;
優選的,步驟(2)的反應溫度50-70℃,反應時間2-3小時。
在本發明的一個實施例中,所述的步驟(1)包括:室溫下,將聚合物微球懸浮于溶劑中,使用硝化試劑對其表面進行硝化處理,使其表面含有硝基官能團,處理后的顆粒洗滌,干燥。
優選的,所述的懸浮用溶劑為二氯甲烷或四氫呋喃;
優選的,所述的洗滌試劑為蒸餾水和/或甲醇。
在本發明的一個優選實施例中,所述的步驟(2)包括:將步驟(1)得到的硝化顆粒懸浮于溶劑中,氮氣保護下,加入飽和氨水和水溶性亞鐵鹽,升溫進行氧化還原反應,反應后的顆粒洗滌,浮選,干燥。
優選的,所述的懸浮用溶劑為蒸餾水;
優選的,所述的洗滌試劑為蒸餾水和/或甲醇。
本發明還提供一種由上述方法制備得到的具有順磁性能的聚合物微球;
優選的,所述的微球為具有順磁性能的多孔交聯聚苯乙烯顆粒;
優選的,所述的微球的鐵含量為1-20%(質量百分數),優選為1-10%,更優選為5-10%。
在本發明的一個優選實施例中,所述的微球的鐵含量為10%;
優選的,所述的微球為球形;
優選的,所述的微球的粒徑為100-400目,更優選為200-400目;
優選的,所述的微球的比表面積100-500m2/g,更優選為300-450m2/g;
優選的,所述的微球的孔徑為3-50nm,更優選為3-10nm。
本發明還提供一種表面環氧基活化的磁性聚合物微球的制備方法,所述方法包括:利用環氧基單體對上述具有順磁性能的聚合物微球表面進行環氧基活化;
優選的,所述的環氧基單體為丙烯酸縮水甘油酯、甲基丙烯酸縮水甘油酯烯丙基縮水甘油酯、丁烯基縮水甘油酯中的一種或多種,和/或,二縮水甘油醚、環氧醇中的一種或多種;
優選的,所述的二縮水甘油醚為丁二醇二縮水甘油醚;
優選的,所述的環氧醇為環氧丙醇。
在本發明的一個優選實施例中,所述的環氧基單體為環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的組合;
在本發明的一個更優選的實施例中,所述的環氧基單體為體積比為1:1:1的環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的組合;
優選的,所述的環氧基單體與具有順磁性能的聚合物微球的重量比例為1:3-2:1;
優選的,所述的的反應溫度為65-75℃;
優選的,所述的的反應時間為16-24h。
在本發明的一個優選實施例中,上述方法包括:
將上述具有順磁性能的聚合物微球懸浮于溶劑中,加入環氧丙醇、甲基丙烯酸縮水甘油醚、丁二醇二縮水甘油醚,攪拌下加熱反應,反應后的顆粒洗滌,干燥。
優選的,所述的懸浮用溶劑為乙二醇二甲醚;
優選的,所述的洗滌試劑為無水乙醇和/或甲醇。
本發明還提供一種上述方法制備得到的表面環氧基活化的磁性聚合物微球。
本發明還提供一種表面功能化的磁性聚合物微球的制備方法,所述方法包括:功能化反應物與上述表面環氧基活化的磁性聚合物微球表面反應;
在本發明的一個實施例中,所述的功能化反應物為含功能化基團的化合物,通過與微球表面環氧基反應連接或聚合,實現表面功能化;
在本發明的一個優選實施例中,所述的功能化反應物為功能化聚合單體,所述方法包括:采用自由基聚合技術,引發功能化聚合單體在上述表面環氧基活化的磁性聚合物微球表面進行聚合反應;
步驟(2)中,采用的引發劑可為偶氮類引發劑、有機或無機過氧化物引發劑,優選自:過硫酸銨、過硫酸鉀、偶氮二異丁腈、過氧化苯甲酰、2,2’-偶氮雙(2-甲基丙脒)二鹽酸鹽;
所述的功能化基團優選自:羥基、羧基、氨基、巰基、酰胺基、季銨基、磺酸基、咪唑、吡啶、吡咯烷酮基中的一種或多種;
優選的,所述的功能化聚合單體含有乙烯基和功能化基團。
在本發明的一個優選實施例中,所述的功能化聚合單體選自:乙烯基吡咯烷酮、甲基丙烯酸羥乙酯、苯乙烯磺酸鈉、乙烯基咪唑、甲基丙酰氧乙基三甲基氯化銨、丙烯酰胺;
優選的,所述的引發劑與功能化聚合單體的重量比為1:10-100,優選為1:20-40;
優選的,所述的功能化聚合單體與步驟(1)得到的顆粒的重量比為1:5-100,優選為1:5-50,更優選為1:5-10。
在本發明的一個優選實施例中,上述方法包括:
將上述表面環氧基活化的磁性聚合物微球懸浮于溶劑中,加入引發劑、功能化聚合單體,升溫聚合,產物洗滌、干燥。
優選的,所述的溶劑選自:乙醇、甲醇、水及各種比例的乙醇(或甲醇)-水混合溶液,所述的溶劑與顆粒的體積重量比為5-20ml/g;
優選的,所述的洗滌試劑選自:無水乙醇、甲醇、水中的一種或多種。
本發明另一方面還提供一種上述方法制備得到的表面功能化的磁性聚合物微球。
優選的,所述的微球為具有順磁性能的多孔交聯聚苯乙烯顆粒;
優選的,所述的微球鐵含量為1-20%(質量百分數),優選為1-10%,更優選為5-10%。
在本發明的一個優選實施例中,所述的微球的鐵含量為10%;
優選的,所述的微球為球形;
優選的,所述的微球的粒徑為100-400目,更優選為200-400目;
優選的,所述的微球的比表面積100-500m2/g,更優選為300-450m2/g;
優選的,所述的微球的孔徑為3-50nm,更優選為3-10nm;
優選的,所述的微球的功能化基團選自:羥基、羧基、氨基、巰基、酰胺基、季銨基、磺酸基、咪唑、吡啶、吡咯烷酮基中的一種或多種。
本發明還提供一種上述制備方法和制備得到的微球在生物工程(如固定化酶)、生物醫學(如靶向藥物、酶標、臨床診斷)、細胞學(如細胞分離、細胞標記)、化學分離(如固相萃取)等領域中的應用。
優選的,所述的應用為在固相萃取中的應用。
本發明還提供一種固相萃取材料,包括上述具有順磁性能的聚合物微球、表面環氧基活化的磁性聚合物微球、表面功能化的磁性聚合物微球;
在本發明的一個優選實施例中,所述的固相萃取材料包括上述表面功能化的磁性多孔交聯聚苯乙烯球形顆粒,所述功能化的基團選自:羥基、吡咯烷酮基、磺酸基、咪唑基、季銨基、酰胺基中的一種或多種;
優選的,所述的微球鐵含量為1-20%,優選為1-10%,更優選為5-10%。
在本發明的一個優選實施例中,所述的微球的鐵含量為10%。
本發明還提供一種上述固相萃取材料的制備方法,所述制備方法包括上述具有順磁性能的聚合物微球的制備步驟;
優選的,所述制備方法還包括利用環氧基單體對上述具有順磁性能的聚合物微球表面進行環氧基活化的步驟;
優選的,所述制備方法還包括功能化反應物與上述表面環氧基活化的磁性聚合物微球表面反應的步驟。
本發明中所述的聚合物微球顆粒的干燥優選使用真空干燥方式。
本發明提供的具有順磁性能的聚合物微球的制備方法,包括以聚合物微球為起始原料,使用硝化試劑對其表面進行硝化,而后通過與水溶性亞鐵鹽的氧化還原反應在微球表面生成磁性材料,得到具有順磁性能的聚合物微球,還可以在此基礎上對上述微球表面進行環氧基活化、功能化衍生,得到表面功能化的磁性聚合物微球。通過優化和控制微球鐵含量,可以盡量減小磁化過程對微球孔徑、比表面積等參數的影響,且后續磁化微球的表面官能化更容易實現,微球的吸附效果更好。上述制備方法工藝簡單,反應條件溫和,工藝可操作性強,選擇性多,易操作,制備效率較高。得到的具有順磁性能的聚合物微球,特別是表面功能化的交聯聚苯乙烯球形顆粒采用外加磁場即可實現快速高效分離,可以再生和重復利用,比表面積大,且表面功能化,可廣泛用于固定化酶、靶向藥物制備、酶或同位素標記、細胞分離、細胞表面標記、固相萃取,特別是用作固相萃取材料的效果好。
附圖說明
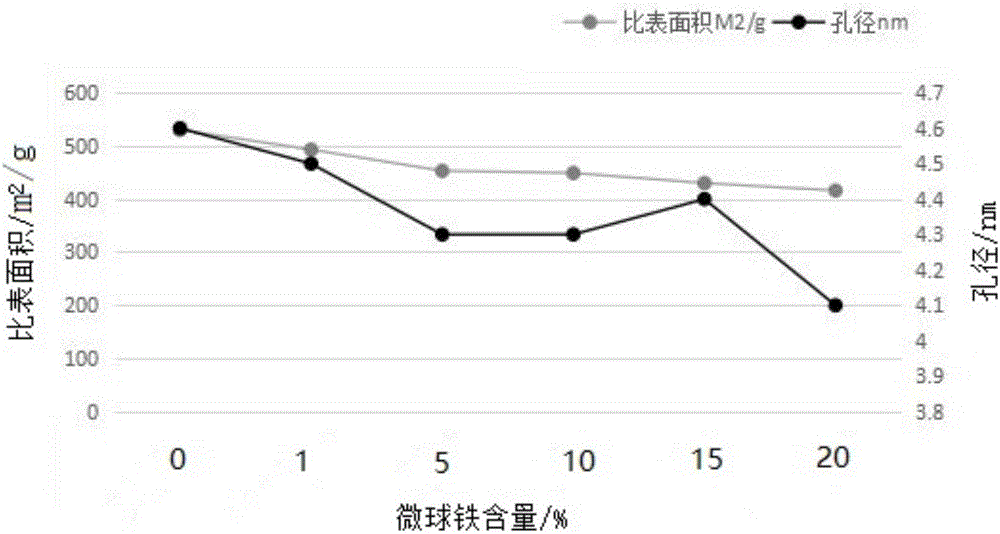
圖1所示為本發明實施例3提供的鐵含量對磁性微球的比表面積、孔徑的影響結果。
圖2所示為本發明實施例4提供的鐵含量對磁性微球的吡咯烷酮取代度的影響結果。
圖3所示為本發明實施例5提供的鐵含量對吡咯烷酮順磁性微球的對乙酰氨基酚吸附率的影響結果。
圖4所示為本發明實施例11提供的磁性微球吸的萃取實驗結果。
具體實施方式
下面將結合本發明實施例中的附圖,對本發明實施例中的技術方案進行清楚、完整地描述,顯然,所描述的實施例僅是本發明一部分實施例,而不是全部的實施例。基于本發明中的實施例,本領域普通技術人員在沒有作出創造性勞動前提下所獲得的所有其他實施例,都屬于本發明保護的范圍。
實施例1
制備步驟如下:
(1)10.0g多孔交聯聚苯乙烯球形顆粒(200-300目,80%交聯,400m2/g)懸浮于100ml二氯甲烷中,加入濃硝酸(65-70wt%)3ml,室溫下攪拌12h。產物過濾,依次用水、甲醇洗滌,60℃真空干燥6小時以上。
(2)將步驟(1)制得的干燥顆粒懸浮于50ml蒸餾水中,氮氣保護下加入40ml飽和氨水、13.0g七水合硫酸亞鐵,攪拌下60℃反應3小時。產物過濾,水洗、浮選,60℃真空干燥6小時以上。
(3)將6.0g步驟(2)制得的產物懸浮于40ml乙二醇二甲醚中,加入環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液(體積比為1:1:1)3.0g,65℃攪拌反應24小時。反應后的顆粒用無水乙醇、甲醇洗滌,真空干燥6小時以上。
(4)將2.0g步驟(3)得到的顆粒懸浮于20ml無水乙醇中,加入乙烯基吡咯烷酮0.20g、0.01g偶氮二異丁腈,攪拌下70℃反應24小時。產物乙醇、甲醇洗滌、真空干燥6小時以上,得到吡咯烷酮功能化的顆粒。制得的產物經測試,具有順磁性,含鐵量6%,200-300目,球形,比表面316m2/g,孔徑4.6nm。
實施例2
制備步驟如下:
(1)10.0g多孔交聯聚苯乙烯球形顆粒(200-400目,60%交聯,500m2/g)懸浮于100ml二氯甲烷中,加入濃硝酸(65-70wt%)冰醋酸的混合液(體積比1:1)4ml,室溫下攪拌12h。產物過濾,依次用水、甲醇洗滌,60℃真空干燥6小時以上。
(2)將步驟(1)制得的干燥顆粒懸浮于50ml蒸餾水中,氮氣保護下加入40ml飽和氨水、9.2g四水合氯化亞鐵,攪拌下75℃反應2小時。產物過濾,水洗、浮選,60℃真空干燥6小時以上。
(3)將6.0g步驟(2)制得的產物懸浮于40ml乙二醇二甲醚中,加入環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液(體積比為1:1:1)3.0g,65℃攪拌反應24小時。反應后的顆粒用無水乙醇、甲醇洗滌,真空干燥6小時以上。
(4)將2.0g步驟(3)得到的顆粒懸浮于20ml無水乙醇中,加入0.2g甲基丙烯酸羥乙酯,0.01g偶氮二異丁腈,攪拌下70℃反應24小時。產物乙醇、甲醇洗滌、真空干燥6小時以上,得到羥基功能化的顆粒。
制得的產物經測試,具有順磁性,含鐵量8%,200-400目,球形,比表面401m2/g,孔徑3.8nm。
實施例3
參照實施例1步驟(1)和(2)制備磁性微球,調節硝化試劑、亞鐵鹽添加量,得到不同鐵含量的磁性微球,以用于磁化的基礎聚合物微球(鐵含量為0)為對照,對比磁化得到的鐵含量對微球的比表面積、孔徑的影響,結果如圖1所示。
由圖1結果可知,磁化過程都會使微球比表面積下降,不同鐵含量對微球的孔徑影響不同,鐵含量為1%和15%,對微球的孔徑影響相對較小。
實施例4
(1)參照實施例1步驟(1)和(2)制備磁性微球,調節硝化試劑、亞鐵鹽添加量,制備得到鐵含量分別為1%、3%、5%、8%、10%、12%、15%、18%、20%的磁性微球。
(2)將6.0g步驟(1)制得的產物懸浮于40ml乙二醇二甲醚中,加入環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液(體積比為1:1:1)3.0g,65℃攪拌反應24小時。反應后的顆粒用無水乙醇、甲醇洗滌,真空干燥6小時以上。
(3)將2.0g步驟(2)得到的顆粒懸浮于20ml無水乙醇中,加入乙烯基吡咯烷酮0.20g、0.01g偶氮二異丁腈,攪拌下70℃反應24小時。產物經乙醇、甲醇洗滌,真空干燥6小時以上,得到吡咯烷酮功能化的顆粒。經元素分析測試含氮量,折算每克磁性微球中吡咯烷酮的毫摩爾數(mmol/g),結果如圖2所示。
由圖2可知,鐵含量越高,制備得到的磁性微球的吡咯烷酮取代度越低,固相萃取過程優選的相應取代度范圍0.5-0.8mmol/g。即優選鐵含量范圍為1-10%,鐵含量越高順磁性越強,因此優選鐵含量為5-10%。
實施例5
(1)取實施例4制備得到的鐵含量分別為1%、3%、5%、8%、10%、12%、15%、18%、20%的吡咯烷酮順磁性微球各10mg。
(2)將0.30mg對乙酰氨基酚溶解于30ml去離子水并置于50ml離心管中,然后將步驟1中的磁性微球,渦旋振蕩3min,用磁鐵吸附磁性微球,取離心管內液體用HPLC分析,計算HPLC分析數據后,對乙酰氨基酚吸附率與鐵含量關系曲線如圖3所示。
由圖3可知,鐵含量為1-10%的微球吸附效果較好。
實施例6
制備步驟如下:
使用200-400目的多孔交聯聚苯乙烯球形顆粒,采用類似實施例1中的方法得到經環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液反應過的干燥顆粒。取10.0g懸浮于90ml甲醇-水(4:1,體積比)中,加入2.0g苯乙烯磺酸鈉、0.10g過硫酸銨,60℃反應24小時,得到磺酸基功能化的顆粒。
產物經洗滌干燥后測試具有順磁性,比表面384m2/g,孔徑4.5nm。
實施例7
使用200-400目的多孔交聯聚苯乙烯球形顆粒,采用類似實施例1中的方法得到經環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液反應過的干燥顆粒。取15.0g懸浮于120ml乙醇中,加入1.5g乙烯基咪唑、0.075g過氧化苯甲酰,75℃反應18小時,得到咪唑功能化的顆粒。
產物經洗滌干燥后測試具有順磁性,比表面384m2/g,孔徑4.5nm。
實施例8
使用200-400目的多孔交聯聚苯乙烯球形顆粒,采用類似實施例1中的方法得到經環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液反應過的干燥顆粒。取10.0g懸浮于100ml乙醇-水(9:1,體積比)中,加入1.5g甲基丙酰氧乙基三甲基氯化銨,0.05g2,2’-偶氮雙(2-甲基丙脒)二鹽酸鹽,70℃反應20小時,得到0.5-0.7mmol/g季銨基功能化的顆粒。
產物經洗滌干燥后測試具有順磁性,比表面413m2/g,孔徑4.5nm。
實施例9
使用200-400目的多孔交聯聚苯乙烯球形顆粒,采用類似實施例1中的方法得到經環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液反應過的干燥顆粒。取10.0g懸浮于100ml乙醇-水(3:1,體積比)中,加入2.0g丙烯酰胺、0.075g過氧化苯甲酰,75℃反應24小時,得到酰胺功能化的顆粒。
產物經洗滌干燥后測試具有順磁性,比表面384m2/g,孔徑4.5nm。
實施例10
制備步驟如下:
(1)Fe3O4納米粒子制備:在N2環境下,將一定量、離子數比n(Fe3+):n(Fe2+)為2∶1的混合鹽溶液置于三口燒瓶中,加水稀釋,加熱到55℃,緩慢加入過量的氨水反應0.5h,升溫至70℃熟化0.5h,得到黑色產物,洗滌干燥后配成一定濃度備用。
(2)Fe3O4納米粒子表面:改性取3mL硅烷偶聯劑KH-570于圓底燒瓶中,依次加入水、冰醋酸,室溫下攪拌至體系均勻。70℃、N2氛圍下,在上述體系中加入一定量納米Fe3O4水溶液,反應至黑亮的油狀物析出。
(3)聚苯乙烯磁性微球制備:取一定量硅烷化改性的Fe3O4、St、MMA、去離子水于三口燒瓶,超聲分散預處理10min,形成穩定體系,氮氣保護,70℃恒溫水浴,攪拌、冷凝回流,逐滴加入K2S2O8溶液,反應4h,分離洗滌,用1mol/L鹽酸浸泡24h后水洗至中性,磁分離得到聚苯乙烯磁性微球。
(4)使用上述制備的多孔交聯聚苯乙烯球形顆粒,采用類似實施例1中的方法得到經環氧丙醇、甲基丙烯酸縮水甘油酯、丁二醇二縮水甘油醚的混合溶液反應過的干燥顆粒。取10.0g懸浮于100ml乙醇-水(9:1,體積比)中,加入1.5g甲基丙酰氧乙基三甲基氯化銨,0.05g2,2’-偶氮雙(2-甲基丙脒)二鹽酸鹽,70℃反應20小時,得到0.3mmol/g季銨基功能化的顆粒。
產物經洗滌干燥后測試具有順磁性,比表面430m2/g,孔徑4.6nm。
對比實施例8和10可知,采用本申請的方法制備得到的磁性微球更容易實現后續的表面功能化。
實施例11萃取效果實驗
以實施例8中得到的磁性微球做萃取實驗,10mg的多孔季銨基功能化聚苯乙烯磁性微球作為吸附劑、100mL的10mg/L的甲基橙溶液,取8份10mL甲基橙溶液(10mg/L),用0.1mol/L的HNO3和0.1mol/L NaOH調節pH=3、4、5、6、7、8、9、10,分別加入10mg多孔季銨基功能化聚苯乙烯磁性微球吸附劑,經渦旋振蕩然后用磁鐵進行分離,用紫外分光光度計進行測定吸光度值。實驗結果如圖4所示。
以上所述僅為本發明的較佳實施例而已,并不用以限制本發明,凡在本發明的精神和原則之內,所作的任何修改、等同替換等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!