一種基于層析原理的高效分離裝置的制作方法
本發明涉及有機化學合成分離技術領域,特別涉及一種基于層析原理的高效分離裝置。
背景技術:
柱層析技術也稱柱色譜技術。在柱層析技術中,向一根柱子里填充層析硅膠形成固定相,將混合樣品加到柱子上后用特別的溶劑洗脫,溶劑組成流動相。在樣品從柱子上洗脫下來的過程中,根據樣品混合物中各組分在固定相和流動相中的分配系數不同,經過多次反復分配,樣品的不同組分將逐一分離。
柱層析色譜柱的填裝主要有濕法和干法兩種。即濕法裝柱和干法裝柱,二者各有優劣。不論干法還是濕法,硅膠(固定相)的上表面一定要平整,并且硅膠(固定相)的高度一般為15cm左右,太短了可能分離效果不好,太長了也會由于擴散或拖尾導致分離效果不好。柱層析關鍵在于柱子是否裝好和淋洗劑是否選擇恰當。而淋洗劑的選擇則是通過薄層層析(TLC)確定。TLC的作用除了跟蹤反應進程,檢測試劑和原料純度外,一個重要的用途就是為柱層析選擇適當的淋洗劑。 一般的規律為:1、如果比移值(Rf值,指在流動相作用下,原點組分的移動距離與流動相的移動距離之間的比值)越小,則點越晚出來,浪費的溶劑越多。 2、如果Rf提高,則相應接收瓶的容量應小一些。3、理論上Rf合適的高度,應該是在TLC中兩點之間距離剛好可以容納第三點。一般為了得到更好的分離,通常把樣品的Rf值調到0.2-0.3之間,不過如果接收瓶小的話,即使Rf值在0.5,0.6,也能把緊挨著的組分點分開。4、上樣高度越薄,則其擴散程度越低,分離效果越好。建議高度不超過2-3mm。所以,盡量采用粗柱子。5、硅膠高度越長,則浪費的溶劑也越多,不過,接收瓶容量可以更大一些。6、硅膠高度越短,接收瓶容量應該小一些。7、硅膠的高度,比TLC的高度高一些即可8、層析柱一定要致密緊實,無氣泡。柱層析時,棉花塞得太緊,影響洗脫液的流速。
然而,柱層析方法的缺點就是存在技術難度大和分離周期長等問題。對于初學者來說需要較長一段時間才能掌握,即使是操作熟練的提純人員,也往往會由于操作不當而造成分離失敗。比如裝柱不均勻,上樣不整齊, 出現氣泡, 縫隙等會明顯影響分離效率。此外遇到兩種組分Rf值相差很小時,由于找不到合適的流動相而容易分不開。柱層析重裝柱、上樣、層析以及濃縮合并樣品的周期比較長,溶劑消耗量大,因而造成分離效率低下,造成研發周期長。
另一方面,薄層色譜(TLC)也是色譜法中的一種,是快速分離和定性分析少量物質的一種重要實驗技術,屬固—液吸附色譜,兼備了柱色譜和紙色譜的優點,一方面適用于少量樣品(幾到幾微克,甚至0.01微克)的分離;另一方面在制作薄層板時,把吸附層加厚加大,因此,又可用來精制樣品,此法特別適用于揮發性較小或較高溫度易發生變化而不能用氣相色譜分析的質.是將吸附劑、載體或其他活性物質均勻涂鋪在平面板(如玻璃板、塑料片、金屬片等)上,形成薄層(常用厚度為0.25毫米左右)后,在此薄層上進行層析分離的分析方法。TLC具有操作方便、設備簡單、顯色容易等特點,同時展開速率快,一般僅需15~20分鐘;混合物易分離,分辨力一般比以往的紙層析高10~100倍,它既適用于只有0.01μg的樣品分離,又能分離大于500mg的樣品作制備用,而且還可以使用如濃硫酸、濃鹽酸之類的腐蝕性顯色劑。但是其缺點在于分離量有限,而且塔板數有限而不能連續層析。另外薄層板則成本比較高,不宜用于制備。
技術實現要素:
鑒于以上柱層析方法存在的技術難度大、分離周期長以及溶劑消耗量大等問題,以及薄層層析方法的分離量小以及不能連續層析等缺點;本發明的目的在于提供一種基于層析原理的高效分離裝置,有機結合柱層析方法和薄層層析方法的優點,揚長避短,從而有效地提高了混合物的層析分離效果。
本發明一種基于層析原理的高效分離裝置,包括層析載體單元和餾分收集單元;
所述層析載體單元為五邊形層析復合板,包括前板、間隔墊、后板、過濾墊層;所述前板、后板的形狀均為關于豎軸對稱的倒五邊形,所述間隔墊設置于所述前板、后板之間,沿所述前板、后板周邊布置;所述前板、后板、間隔墊共同形成僅上部開口的腔體;所述過濾墊層設置于所述腔體下部;
所述餾分收集單元包括餾分收集瓶、第一導流細管;所述第一導流細管從所述層析載體單元底部引出,與所述餾分收集瓶連通。
進一步的,所述餾分收集單元還包括膠塞、第二導流細管、橡膠管、抽真空裝置;所述膠塞設置于所述餾分收集瓶上口,所述第二導流細管一端穿過所述膠塞與所述餾分收集瓶連通,另一端通過所述橡膠管與所述抽真空裝置連接。
進一步的,所述后板在上部長于前板,所述后板長出所述前板的部分用以引流。
進一步的,所述前板、后板的厚度為2mm-10mm;所述前板、后板之間的間隔為1.0mm-20mm。
進一步的,所述前板、后板的材料為耐有機溶劑的材料。
進一步的,所述前板、后板的材料為透明或半透明材料;所述透明或半透明材料為玻璃、石英、高密度聚乙烯、聚丙烯或PET。
進一步的,所述分離裝置所用的固定相為正相填料或反相填料,所述正相填料為硅膠、CN(腈基)鍵合硅膠、DIOL(二醇基)鍵合硅膠、NH2(氨基)鍵合硅膠、氨基取代填料,硅酸鎂填料,氧化鋁填料、大孔樹脂、離子交換樹脂中的任一種;所述反相填料為 C18(十八烷基)鍵合硅膠、C8(辛烷基)鍵合硅膠、C2(乙基)鍵合硅膠、CH(環已烷基)鍵合硅膠、PH(苯基)鍵合硅膠中的任一種。
進一步的,所述分離裝置所用的流動相為有機溶劑。
進一步的,所述有機溶劑為醋酸、乙腈、水、甲醇、乙醇、異丙醇、丙酮、丁酮、環己酮、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙醚、石油醚、環氧丙烷、二氯甲烷、氯仿、四氯化碳、氯苯、苯、甲苯、二甲苯、環己烷、正己烷等脂肪烷中的任一種或者混合溶劑。
進一步的,所述餾分收集單元由夾持固定配件固定于鐵架或網格架上,所述夾持固定配件為便攜性鐵夾或燒瓶夾。
本發明的有益效果為:
1.有機結合了柱層析和薄層層析的特點。
2.分離量大、連續層析,而且分離效果好。
3.分離速度快,明顯優于制備板的分離速度。
4.提高了分離效率,節省了大量層析所用溶劑。
5.當使用透明板或半透明板時,可以直接實時觀察層析餾分走動情況,減省重復用TLC跟蹤工作,還可以及時針對性收集和跟換收集瓶,提高了工作效率。
6.可以連接真空減壓裝置,進一步的縮短樣品分離時間。
附圖說明
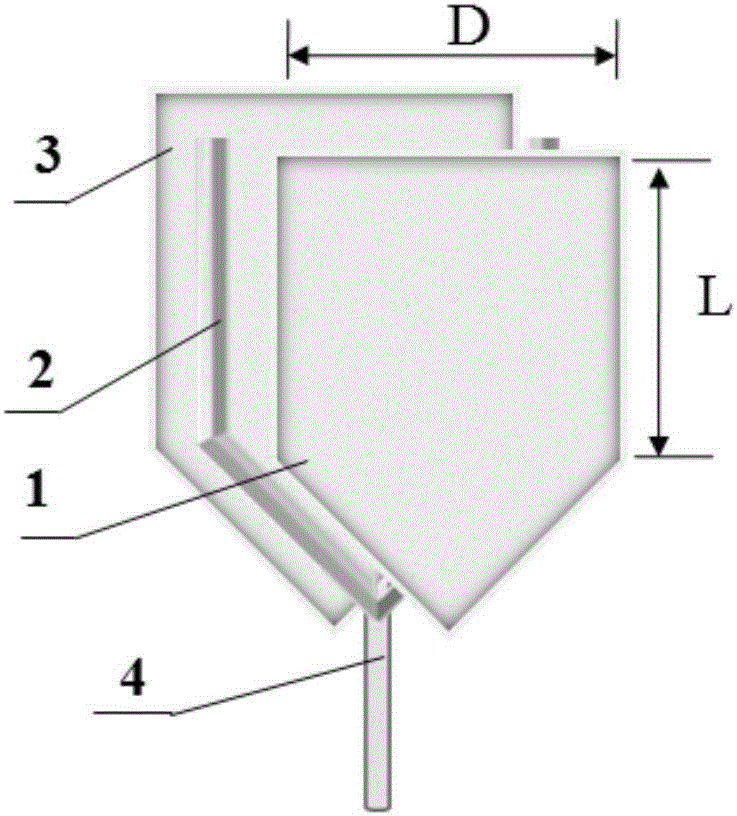
圖1所示為本發明實施例中的五邊形層析復合板的結構示意圖。
圖2所示為本發明實施例一種基于層析原理的高效分離裝置結構示意圖。
圖中:1-前板、2-間隔墊、3-后板、4-第一導流細管、5-過濾墊層、6-膠塞、7-餾分收集瓶、8-鐵架或網格架、9-夾持固定配件、10-第二導流細管、11-橡膠管、12-抽真空裝置、13-固定相。
具體實施方式
下文將結合具體附圖詳細描述本發明具體實施例。應當注意的是,下述實施例中描述的技術特征或者技術特征的組合不應當被認為是孤立的,它們可以被相互組合從而達到更好的技術效果。在下述實施例的附圖中,各附圖所出現的相同標號代表相同的特征或者部件,可應用于不同實施例中。
本發明實施例一種基于層析原理的高效分離裝置,包括層析載體單元和餾分收集單元;層析載體單元為層析分離混合物的場所;餾分收集單元用于接收層析過程中的餾分;兩者通過第一導流細管4及膠塞連接。另外,為了提高流動相的流動速度,可以再連減壓裝置(抽真空裝置12);所述餾分收集單元可以由夾持固定配件9固定于鐵架或網格架8上,所述夾持固定配件9為便攜性鐵夾或燒瓶夾,優選寬度大于2cm。
所述層析載體單元為五邊形層析復合板,包括前板1、間隔墊2、后板3、過濾墊層5;所述前板1、后板3的形狀均為關于豎軸對稱的倒五邊形,所述間隔墊2設置于所述前板1、后板3之間,沿所述前板1、后板3周邊布置;所述前板1、后板3、間隔墊2共同形成僅上部開口的腔體;所述過濾墊層5設置于所述腔體下部;
所述餾分收集單元包括餾分收集瓶7、第一導流細管4;所述第一導流細管4從所述層析載體單元底部引出,與所述餾分收集瓶7連通。
當需要配備減壓裝置以提高分離效率時,所述餾分收集單元還可以包括膠塞6、第二導流細管10、橡膠管11、抽真空裝置12;所述膠塞6設置于所述餾分收集瓶7上口,所述第二導流細管10一端穿過所述膠塞6與所述餾分收集瓶7連通,另一端通過所述橡膠管11與所述抽真空裝置12連接。
優選的,所述后板3在上部略長于前板1,所述后板3長出所述前板1的部分用以引流。所述前板1、后板3的厚度分別為0.1mm-10mm;所述前板1、后板3之間的間隔為0.1mm-20mm。所述上板1、下板3的尺寸可根據分離量確定,一般長度L不能小于寬度D,,比如20cm*30cm;
所述前板1、后板3的材料為耐有機溶劑的材料;優選的,所述前板1、后板3的材料為透明或半透明材料;具體可以為玻璃、石英、高密度聚乙烯、聚丙烯或PET。
所述分離裝置所用的固定相13為正相填料或反相填料,所述正相填料為硅膠、CN(腈基)鍵合硅膠、DIOL(二醇基)鍵合硅膠、NH2(氨基)鍵合硅膠、氨基取代填料,硅酸鎂填料,氧化鋁填料、大孔樹脂、離子交換樹脂中的任一種;所述反相填料為 C18(十八烷基)鍵合硅膠、C8(辛烷基)鍵合硅膠、C2(乙基)鍵合硅膠、CH(環已烷基)鍵合硅膠、PH(苯基)鍵合硅膠中的任一種。
所述分離裝置所用的流動相可以為各種有機溶劑;優選的,所述有機溶劑為醋酸、乙腈、水、甲醇、乙醇、異丙醇、丙酮、丁酮、環己酮、乙酸甲酯、乙酸乙酯、乙酸丙酯、乙醚、石油醚、環氧丙烷、二氯甲烷、氯仿、四氯化碳、氯苯、苯、甲苯、二甲苯、環己烷、正己烷等脂肪烷中的任一種或者混合溶劑。
在實際使用中,層析復合板下端的流動相在重力作用下自發地沿斜邊流至下頂點,再經過第一導流細管4排出層析單元,這樣能保證層析出的餾分能夠及時順序地流出以保證層析的分離效果。
五邊形層析復合板的填裝方法和普通柱層析的填裝方法一樣,也有濕法和干法兩種。在使用時,需要在層析復合板的下頂點處填充一些棉花(過濾墊層5)用以擋住層析體內的固定相13不被流動相沖走。濕法裝柱是先把固定相13用適當的溶劑拌勻后,再用楔形漏斗緩緩填入五邊形層析復合板,然后再用大量的淋洗劑沖洗復合板或者連接減壓裝置等方式以把固定相13壓實。干法裝柱則是直接往層析復合板里填入固定相13,然后再輕輕敲打柱子兩側至固定相界面不再下降為止,然后再填入固定相13至合適高度,當固定相13上表面不平整時,可以通過輕敲打復合板前后兩側或者用鐵絲輕輕攪動固定相13上表面,以獲得較平整的上表面。最后再用抽真空裝置12直接抽,這樣就會使得柱子裝的很結實。接著是用淋洗劑淋洗層析復合板,一般淋洗劑是采用TLC分析得到的展開劑的比例再稀釋一倍后的溶劑。可以上面加壓,下面再用油泵抽,這樣可以加快速度。雖然干法裝柱較方便,但由于溶劑和固定相之間的吸附放熱,容易產生氣泡,特別是在使用如乙醚,二氯甲烷等低沸點的淋洗劑時更為明顯。解決的辦法是首先固定相13一定要天結實;然后是一定要用較多的淋洗劑淋洗,一定要到柱子的下端不再發燙,恢復到室溫后再撤去壓力。
五邊形層析復合板的上樣方法也可以采用干法或濕法:干法上樣就是把待分離的樣品用少量溶劑溶解后,再加入1. 2-1.5倍固定相,拌勻后旋去溶劑,將得到的粉末加到柱子頂層。注意旋干后,不能看到明顯的樣品固體顆粒,如果有明顯的樣品顆粒,說明加入的固定相13不夠而需要補加固定相13。干法上樣操作麻煩,但可以保證樣品層平整度。濕法上樣則是將液體或溶液樣品直接加在填料的表面。且對于液體樣品或粘稠狀半固體樣品只能采用濕法上樣。用少量溶劑(最好就是展開劑,如果展開劑的溶解度不好,則可以用一極性較大的溶劑,但必須少量)將樣品溶解后,再用膠頭滴管轉移得到的溶液,沿著層析柱內壁均勻加入。然后用少量溶劑洗滌后再加入。濕法較方便,熟手一般采用此法。總之,不管是干法或濕法裝柱都要求盡可能薄的平鋪在固定相上,太厚會造成分離效果差或分不開樣品。
柱色譜上樣與柱填料量的最佳比值也可通過薄層色潛實驗確定。首先確定最佳上樣液濃度。在薄層色譜中以能夠不使樣品點因過載而嚴重拖尾導致分離失敗,這樣的最高濃度為最佳上樣液濃度。薄層色譜扳上的有效固定相體積的計算方式為:V=長×寬×厚,其中長為基線到溶劑前沿的長度,寬為樣品點的最大寬度,厚為薄層色譜板的固定相厚度。通常固定相13填料的平均密度為0.5g/cm3。最后由最佳點樣質量和有效固定相體積,計算出樣品質量與固定相13質量的最佳比值。最后轉化為五邊形復合層析的色譜條件,也就是樣品質量與固定相質量的比值保持不變。
當樣品在填滿層析固定相13的層析復合板上樣以后,淋洗劑在重力作用下開始洗脫固定相上的組分。和普通層析一樣,由于混合物中不同成分的極性存在差異,則與固定相13的作用力不一樣,與固定相13作用力強的組分在層析過程中被洗脫的速度較慢,而作用力弱的組分則容易被洗脫而走在前面。如果以硅膠為例,則極性小的組分由于速度較快而率先餾出,而極性大物質由于速度較慢而后餾出來,從而實現對樣品混合物進行分離。
本發明實施例分離裝置由于復合板上方沒有密封,所以不能在加壓條件下收集餾分,但是可以在常壓和減壓下收集餾分。本發明中層析復合板如果使用透明或半透明的有機或無機材料,當分離有色樣品時,層析過程中可以直接在復合板上觀察層析餾分實時走動情況。當分離無色樣品時,可以在固定相13中加入熒光粉,在紫外燈的幫助下一樣可以直接在復合板上觀察層析餾分實時走動情況。由于省去了大量的TLC跟蹤點板工作,提高了工作效率。
由于本發明中復合板上面可以不斷加入流動相,而下方可以連續不斷地導流出餾分,因而可以實現連續層析。固定相層較薄,增加了層析的塔板數,分離效果好,相當于薄層層析。相對柱層析來說,復合板的寬度較寬,相當于并聯了若干個細長的層析柱,在分離效果、分離量以及分離時間上明顯優于薄層層析和柱層析。
實施例
在本實施例中,提供一種采用前述的分離裝置在減壓條件下分離的方法。
首先安裝和固定分離裝置:將20cm*25cm的五邊形層析復合板垂直固定在鐵架或網格架8上,其下頂點通過金屬導流細管針(9mm)(第一導流細管4)和反口橡膠塞(24#)(膠塞6)與100mL磨口三角瓶(24#)(餾分收集瓶7)相連,同時在橡膠塞中插入金屬細管針(9mm)(第二導流細管10)連接真空泵(抽真空裝置12),借助細鐵絲將脫脂棉(過濾墊層5)攝入五邊形層析復合板的下頂點處以堵住金屬導流細管口。
濕法裝柱:把200-300目硅膠(13)用石油醚拌勻成稀漿糊狀,經楔形漏斗緩緩填入五邊形層析復合板,待高度達到約15cm左右時停止加入硅膠。然后用石油醚沖洗復合板或者連接減壓裝置不加載樣品淋洗3-5次,以便把硅膠層壓實。在此過程如果發現硅膠上表面不平整,可以輕敲復合板或者用細鐵絲撥弄硅膠表面以令平整。
干法上樣:將待分離的有色樣品(便于觀察層析效果)用少量二氯甲烷溶解后,再加入1. 2-1.5倍樣品的硅膠,拌勻后旋去溶劑以得到粉末。然后用藥勺經楔形漏斗緩緩加入到硅膠上表面,然后用石油醚將殘余在樣品瓶壁、層析復合板壁以及藥勺上的硅膠等也小心沖導硅膠上表面。在此過程中也同樣需要保證上樣后的硅膠上表面具有一定的平整度。
層析和收集:在硅膠層上方緩緩加入淋洗劑(石油醚:二氯甲烷=1:1),層析板下方的三角瓶每30mL-50mL更換一次三角收集瓶,以便及時分開層析下來的餾分。此過程中可以間斷的開啟抽真空裝置12以提高層析速度。在開啟過程中,注意在關閉抽真空裝置12后,層析系統里面還有一定的負壓。所以為了穩定更換三角收集瓶,需要注意在更換操作前一段時間關閉減壓裝置。
本發明實施例分離裝置在分離0.5克有色樣品時具有良好的分離效果,僅需要1個小時就能完成層析分離操作,而且樣品層析明顯,共消耗1000ml淋洗劑(V石油醚:V二氯甲烷=1:1)。而相同的柱層析雖然也能較好的分離樣品,但是耗時2個小時左右,需要用至少1400ml的淋洗劑(V石油醚:V二氯甲烷=1:1)。
本發明的有益效果為:
1.有機結合了柱層析和薄層層析的特點。
2.分離量大、連續層析,而且分離效果好。
3.分離速度快,明顯優于制備板的分離速度。
4.提高了分離效率,節省了大量層析所用溶劑。
5.當使用透明板或半透明板時,可以直接實時觀察層析餾分走動情況,減省重復用TLC跟蹤工作,還可以及時針對性收集和跟換收集瓶,提高了工作效率。
6.可以連接真空減壓裝置,進一步的縮短樣品分離時間。
本文雖然已經給出了本發明的幾個實施例,但是本領域的技術人員應當理解,在不脫離本發明精神的情況下,可以對本文的實施例進行改變。上述實施例只是示例性的,不應以本文的實施例作為本發明權利范圍的限定。
- 還沒有人留言評論。精彩留言會獲得點贊!